אמיוטרופיה של עמוד השדרה (ניוון שרירי עמוד השדרה או SMA) היא הפרעה חשוכת מרפא, כמעט תמיד תורשתית הנגרמת על ידי מוטציה גנטית בכרומוזום 5.
מוטציה בגן CMA מובילה למחסור בחלבון, הנחוץ לבניית מבני RNA חלבוני, ולהתפתחות לא מספקת של שרירים מפוספסים, בעיקר גפיים תחתונות, ו צוואר הרחםוראשים.
המחלה יכולה להתבטא מרגע הלידה, ואפילו כשהעובר עדיין ברחם, ובכל זמן בחיים. ביילודים, אמיוטרופיה של עמוד השדרה מובילה לרוב למוות מוקדם, אך במקרים מסוימים היא יכולה להיות קלה, בעיקר בגיל מבוגר. הבה נבחן ביתר פירוט את התכונות של פתולוגיה זו.
אמיוטרופיה של עמוד השדרה - הכל על המחלה
היסטוריה וסטטיסטיקה
SMA זה מספיק מחלה נדירה, פתוח רופא גרמני Werdnig בשנת 1891. אחד מכל 6-10 אלף אנשים חולה בו, אבל כל אדם 50 הוא נשא של הגן SMA רצסיבי.
בשנת 1898, ורדניג ומדען נוסף הופמן קבעו שהגורם ל-SMA הוא נגע ניווני ומספר לא מספיק של נוירונים מוטוריים (מוטוריים) של הקרניים הקדמיות עמוד שדרה- SMN (נוירונים מוטוריים הישרדותיים).

כבר במאה ה-20 (בשנת 1956) גילו מדענים אחרים קוגלברג וולנדר צורה פחות ממאירה ומתונה יותר של SMA, המשפיעה על צעירים ומבוגרים.
סוג תורשה באמיוטרופיה של עמוד השדרה
המחלה יכולה לעבור בתורשה מכל אחד מהסוגים:
- דומיננטי אוטוזומלי;
- אוטוזומלית רצסיבית;
- דומיננטי מקושר X;
- X-linked רצסיבי.
בהקשר זה, זה מסווג הרבה צורות שונות SMA.
צורת הילדות של SMA עוברת בתורשה בצורה אוטוזומלית רצסיבית: אם שני ההורים נשאים, אז רביעית מהצאצאים שלהם יחלה.
אופן התורשה האוטוזומלי הדומיננטי של SMA מוביל לביטוי המחלה בילדים בסבירות של 50%, גם אם רק הורה אחד מושפע.
סוגי אמיוטרופיה של עמוד השדרה
ניוון שרירי עמוד השדרה מתחלק לארבע צורות:
- Infantile (I) - Werdnig-Hoffmann ניוון שרירי עמוד השדרה: מאובחנת מלידה ועד שישה חודשים.
- בינוני (II) - מחלת דובוביץ: משבעה חודשים עד שנה וחצי.
- נוער (III) - ב. קוגלברג-וולנדר: אחרי שנה וחצי.
- מבוגר (IV): לאחר 35 שנים.
תסמינים של ניוון שרירי עמוד השדרה
תסמינים נפוצים ב-SMA:
- נזק לשרירים הפרוקסימליים (האמצעיים) ולפאשיה;
- שימור הרגישות ברובם מקרים קליניים;
- פיגור שכלי ו התפתחות נפשיתעם amyotrophy שרירי עמוד השדרה הם נדירים ביותר;
- אפשרי עם כמה סוגים של ניוון הוא לא רק את השרירים של הגפיים, אבל הנשימה, לעיסה ובליעה.

חומרת SMA
- החמור והבלתי חיובי ביותר הוא SMA מהסוג הראשון (אינפנטילי) - Werdnig-Hoffman spinal amyotrophy, בה ילדים אינם מסוגלים לבצע תנועות אקטיביות, להחזיק את הראש ולשבת בכוחות עצמם. האכלה ניתנת לתינוק בקושי רב, שכן קשה לו לינוק חלב ולבלוע אותו.
- מחלת דובוביץ' (צורת ביניים II של SMA) פחות ממאירה: איתה ילדים מסוגלים לשבת, להחזיק את הראש ולאכול, אך עדיין אינם מסוגלים ללכת.
- הפחות חמורה היא צורת הנעורים: למרות חולשת השרירים, הילד מסוגל ללמוד ללכת, אך המחלה, אם כי לאט, מתקדמת ועלולה להוביל לנכות מוקדמת.
- רביעי צורה למבוגריםעקב החולשה של החלקים הפרוקסימליים m-c, SMA יכול להוביל לחוסר אפשרות של תנועה עצמאית, אובדן רפלקסים, אך הפרוגנוזה ביחס לתוחלת החיים נשארת חיובית.

סוגים אחרים של ניוון שרירי עמוד השדרה
בנוסף למוטציות בגנים, גורם לתבוסהשרירים פרוקסימליים, יש פתולוגיות דומות, עם סוג אחרתורשה, המובילה לאטרופיה של השרירים והפאשיה של הדיסטלי (קטעים סופניים).
הרשימה שלהם די גדולה, בואו נסכם את המחלות בטבלה קטנה:
| שם SMA | סוג ירושה | תכונות ותסמינים |
| SMAX1 | X-linked רצסיבי | זה נצפה בעיקר אצל קשישים, משפיע על העצבים הבולבריים של הגולגולת, גורם לשיתוק יורד. |
| SMAX2 | X - קלאץ'. רצסיבי | מִלֵדָה צורה אגרסיביתמוביל למוות עד 3 חודשים. גורם לחולשה, ארפלקסיה, התכווצויות ושברים. |
| SMAX3 | X - קלאץ'. רצסיבי | זה משפיע בעיקר על בנים. אטרופיה של כל השרירים הדיסטליים. הופעה איטית של סימפטומים |
| DSMA1 דיסטלי | אוטוזומלית רצסיבית | מולד, משפיע בעיקר על הידיים, יתכנו הפרעות נשימה קשות |
| צורות דיסטליות DSMA2 - DSMA5 | אוטוזומלית רצסיבית | כל ארבע הצורות מאופיינות בהתקדמות איטית, DSMA5 מאובחן בצעירים. |
| SMA דיסטלי משני סוגים: VA ו-VB (DSMAVA ו-DSMAVB) | דומיננטי אוטוזומלי | הגפיים העליונות מנוונות בעיקר. |
| DSMA מסוג 2D | אוטוזומלית רצסיבית | נוער ו מחלת מבוגריםעם התפתחות איטית: שרירים פרוקסימליים ודיסטליים מושפעים - תחילה ברגליים, ואז בידיים. |
| DSMA סוג 7A | דומיננטי אוטוזומלי | צורה בוגרת נדירה מאוד עם פגיעה במיתרי הקול. |
| DSMA סוג 2A | דומיננטי אוטוזומלי | מגוון מחלות שארקוט (סוג אללי) |
| SMA לנוער (סוג HMN1) | דומיננטי אוטוזומלי | מתרחש בנוער |
| אמיוטרופיה מולדת של עמוד השדרה | דומיננטי אוטוזומלי | הפרה של עצבוב וניוון של m - ירכיים, רגליים, ברכיים עם התכווצות ועיוות; לפעמים מיתרי הקול מושפעים. |
| ה-SMA של פינקל | דומיננטי אוטוזומלי | היא מתחילה בעיקר בגיל 35-37, אך מקרים של המחלה נרשמו גם בילדות. לאט לאט מתפתח תחילה ברגליים ולאחר מכן בזרועות. הפעילות והרפלקסים מופחתים, רעד לא רצוני (פאסיקולציה) נצפה. |
| SMA Jokel | דומיננטי אוטוזומלי | מושפע אצל מבוגרים פרוקס. ומרוחקת מ - צי. |
| SMA (סוג LED1) | דומיננטי אוטוזומלי | אטרופיה של הגפיים התחתונות ביילודים. |
| PME מסוג SMA | אוטוזומלית רצסיבית | אטרופיה של השרירים הדיסטליים עם פגיעה בעצבוב והתקפים אפילפטיים |
| SMA עם שברי עצם מולדים | אוטוזומלית רצסיבית | תסמינים חמורים, כמו במחלה. ורדניג-הופמן, עמוס שברים. |
| SMA עם היפופלזיה | דומיננטי אוטוזומלי | אנומליה מוחית מולדת עם תסמינים מוחיים, מיקרוצפליה ועיכוב התפתחותי. |
| סוג אסימטרי לנוער SMA | -------------- | זה משפיע על גברים הודים צעירים |

בטבלה זו, יש לשים לב לשני הסוגים האחרונים של אמיוטרופיה בעמוד השדרה:
- SMA עם היפופלזיה מלווה בסטיות בהתפתחות הנפשית והנפשית, שאינה אופיינית לסוגים אחרים של המחלה.
- אמיוטרופיה צעירה אסימטרית (הודית) אינה עוברת בתורשה. במקרה זה, המחלה יכולה להתייצב לאחר שנתיים עד חמש שנים של מהלך איטי. תסמינים של פאסיקולציות נדירים בצורה ספציפית זו.
טיפול באמיוטרופיה של עמוד השדרה
זה בלתי אפשרי ביסודו לרפא סוג זה של מחלה, כמו כל פתולוגיה תורשתית המשפיעה על חוט השדרה או המוח. יעילותן של תרופות רבות המשמשות כיום בטיפול באמיוטרופיה לא הוכחה. מהות הטיפול היא הגדלת החלבון המעורב ביצירת נוירונים מוטוריים SMN.
אפשרויות טיפול ב-SMA
אז, התרופות הבאות משמשות בעיקר:
- חומצה ולפרואית;
- נתרן אוקסיבוטיראט;
- nusinersen (תרופה חדשה שהוצגה בארה"ב ב-2016 לטיפול ב-SMA).
טיפול תומך (תרפיה בכושר, עיסוי, פיזיותרפיה), מיוחד דיאטת חלבון, שלוקח בחשבון התוויות נגד אפשריות. מטופלים שנשללה מהם האפשרות לתנועה עצמאית זקוקים לטיפול סוציאלי.
זהו ניוון שרירי עמוד השדרה הממאיר ביותר המתפתח מלידה או ב-1-1.5 השנים הראשונות לחייו של ילד. זה מאופיין על ידי ניוון שרירים מפוזר גובר, מלווה paresis רפוי, מתקדם לפלגיה מלאה. ככלל, אמיוטרופיה של Werdnig-Hoffman משולבת עם עיוותים בעצמות ואנומליות התפתחותיות מולדות. הבסיס האבחוני הוא אנמנזה, בדיקה נוירולוגית, מחקרים אלקטרופיזיולוגיים וטומוגרפיים, ניתוח DNA וחקר המבנה המורפולוגי. רקמת שריר. הטיפול אינו יעיל, שמטרתו לייעל את הטרופיזם של רקמות העצבים והשרירים.
ICD-10
G12.0ניוון שרירי עמוד השדרה אינפנטילי, סוג I [ורדניג-הופמן]

מידע כללי
אמיוטרופיה של Werdnig-Hoffmann היא הגרסה החמורה ביותר של כל ניוון שרירי עמוד השדרה (SMA). השכיחות שלו היא ברמה של מקרה אחד לכל 6-10 אלף יילודים. כל אדם 50 הוא נשא של גן שונה הגורם להופעת מחלה. אבל בגלל הסוג האוטוזומלי הרצסיבי של תורשה, הפתולוגיה אצל ילד מתבטאת רק כאשר הסטייה הגנטית המתאימה קיימת הן אצל האם והן אצל האב. ההסתברות ללדת ילד עם פתולוגיה במצב כזה היא 25%.
למחלה מספר צורות: מולדת, בינונית (ילדות מוקדמת) ומאוחרת. מספר מומחים מייחדים את הצורה האחרונה כנוסולוגיה עצמאית - אמיוטרופיה של קוגלברג-וולנדר. היעדר טיפול אטיוטרופי ופתוגני, תוצאה קטלנית מוקדמת מעמיד את הטיפול בחולים עם מחלת ורדניג-הופמן בין התוצאות הגבוהות ביותר. משימות מאתגרותמול נוירולוגיה וילדים מודרניים.
גורם ל
אמיוטרופיה של Werdnig-Hoffmann היא פתולוגיה תורשתית המקודדת על ידי התמוטטות במנגנון הגנטי ברמה של הלוקוס 5q13 של הכרומוזום החמישי. הגן שבו מתרחשות מוטציות נקרא הגן המוטורי המוטורי (SMN), הגן האחראי להישרדותם של נוירונים מוטוריים. ל-95% מהחולים במחלת ורדניג-הופמן יש מחיקה של העותק הטלומרי של הגן הזה. חומרת ה-SMA מתואמת ישירות עם אורך אתר המחיקה והנוכחות הנלווית של שינויים (רקומבינציה) בגנים H4F5, NAIP ו- GTF2H2.
התוצאה של הסטייה של הגן SMN היא תת-התפתחות של הנוירונים המוטוריים של חוט השדרה, הממוקמים בקרניו הקדמיות. התוצאה היא עצבוב לא מספק של השרירים, מה שמוביל לאטרופיה בולטת שלהם עם אובדן כוח השרירים ודעיכה מתקדמת של היכולת לבצע פעולות מוטוריות אקטיביות. הסכנה העיקרית היא חולשת שרירים חזה, ללא השתתפותן של תנועות המספקות תפקוד נשימתי בלתי אפשרי. יחד עם זאת, הספירה החושית נשארת ללא פגע לאורך כל המחלה.
תסמינים של אמיוטרופיה
צורה מולדת(SMA I) מתבטא קלינית לפני גיל 6 חודשים. ברחם, זה יכול לבוא לידי ביטוי בתנועה איטית של העובר. לעתים קרובות, היפוטוניה בשרירים הוא ציין מהימים הראשונים של החיים ומלווה בהכחדה של רפלקסים עמוקים. ילדים בוכים חלש, מוצצים גרוע, לא יכולים להרים את הראש. במקרים מסוימים (עם הופעת תסמינים מאוחרת יותר), הילד לומד להחזיק את הראש ואף לשבת, אך על רקע התפתחות המחלה מיומנויות אלו נעלמו במהירות. מאופיין בהפרעות בולבריות מוקדמות, ירידה ברפלקס הלוע, עוויתות פשקולריות של הלשון.
אמיוטרופיה זו של ורדניג-הופמן משולבת עם אוליגופרניה והפרעות בהיווצרות המנגנון העצם-מפרקי: עיוותים בחזה (חזה בצורת משפך וחזה), עקמומיות של עמוד השדרה (עקמת), התכווצויות מפרקים. למטופלים רבים יש חריגות מולדות אחרות: המנגיומות, הידרוצפלוס, כף רגל, דיספלזיה. מפרקי ירך, קריפטורכיזם וכו'.
מהלך ה-SMA I הוא הממאיר ביותר עם חוסר תנועה וגובר במהירות ופראזיס של שרירי הנשימה. האחרון גורם להתפתחות והתקדמות של כשל נשימתי, שהוא הגורם העיקרי למוות. בקשר להפרת הבליעה, אפשר לזרוק לתוכו מזון כיווני אווירעם התפתחות של דלקת ריאות שאיפה, שיכולה להיות סיבוך קטלני של אמיוטרופיה של עמוד השדרה.
צורת ילדות מוקדמת(SMA II) יוצא לראשונה לאחר גיל 6 חודשים. עד תקופה זו, לילדים יש התפתחות גופנית ונוירו-נפשית מספקת, בהתאם לנורמות הגיל, הם רוכשים את הכישורים להחזיק את הראש, להתהפך, לשבת, לעמוד. אבל ברוב המוחלט של המקרים הקליניים, לילדים אין זמן ללמוד ללכת. בדרך כלל, האמיוטרופיה הזו של ורדניג-הופמן באה לידי ביטוי לאחר הרעלת מזון או מחלה זיהומית חריפה אחרת ממנה סובל ילד.
בתקופה הראשונית, paresis היקפי מתרחשת בגפיים התחתונות. ואז הם התפשטו במהירות לגפיים העליונות ולשרירי הגוף. מתפתחת היפוטוניה שרירית מפושטת, רפלקסים עמוקים דוהים. יש התכווצויות של הגידים, רעד של האצבעות, התכווצויות שרירים לא רצוניות (פאסיקולציות) של הלשון. בשלבים המאוחרים יותר מצטרפים תסמינים בולבריים וכשל נשימתי מתקדם. המהלך איטי יותר מזה של הצורה המולדת של מחלת ורדניג-הופמן. מטופלים יכולים לחיות עד גיל 15.
אמיוטרופיה של קוגלברג-וולנדר(SMA III) - האמיוטרופיה השפירה השפירה ביותר יַלדוּת. מתבטא לאחר שנתיים, במקרים מסוימים בתקופה שבין 15 ל-30 שנים. אין פיגור שכלי הרבה זמןהמטופלים מסוגלים לנוע באופן עצמאי. חלקם חיים עד זקנה בשלה, מבלי לאבד את יכולת השירות העצמי.
אבחון
במונחים אבחוניים, עבור נוירולוג ילדים, גיל הופעת התסמינים הראשונים והדינמיקה של התפתחותם, נתוני מצב נוירולוגי (בעיקר נוכחות של הפרעות תנועהסוג היקפי על רקע רגישות שלמה לחלוטין), נוכחות של חריגות מולדות נלוות ועיוותים בעצמות. אמיוטרופיה מולדת של Werdnig-Hoffmann יכולה להיות מאובחנת על ידי רופא ילודים. אבחון דיפרנציאלימבוצע עם מיופתיות, ניוון שרירים פרוגרסיבי של דושן, טרשת צדדית אמיוטרופית, סירינגומיליה, פוליומיאליטיס, תסמונת ילד איטי, שיתוק מוחין, מחלות מטבוליות.
על מנת לאשר את האבחנה, מתבצעת electroneuromyography - מחקר של המנגנון הנוירו-שרירי, שבגללו מתגלים שינויים אופייניים שאינם כוללים את הסוג השרירי העיקרי של הנגע ומצביעים על הפתולוגיה של הנוירון המוטורי. ניתוח ביוכימידם אינו מגלה עלייה משמעותית בקריאטין פוספוקינאז, האופיינית לניוון שרירים מתקדם. בדיקת MRI או CT של עמוד השדרה מקרים נדיריםלַחֲזוֹת שינויים אטרופייםקרניים קדמיות של חוט השדרה, אך מאפשרות להוציא פתולוגיה אחרת של עמוד השדרה (hematomyelia, מיאליטיס, ציסטה וגידול של חוט השדרה).
האבחנה הסופית של אמיוטרופיה של Werdnig-Hoffmann נקבעת לאחר קבלת נתוני ביופסיית שרירים ומחקרים גנטיים. מחקר מורפולוגיביופסיית שריר חושפת ניוון צרור פתוגנומוני של סיבי שריר עם אזורים מתחלפים של ניוון של מיופיברילים ורקמת שריר ללא שינוי, נוכחות של מיופיברילים היפרטרופיים נפרדים, אזורים של גידולי רקמת חיבור. ניתוח DNA המבוצע על ידי גנטיקאים כולל אבחון ישיר ועקיף. על ידי שימוש ב שיטה ישירהניתן גם לאבחן נשא הטרוזיגוטי של סטיית גנים, החשובה בייעוץ גנטי של אחים (אחים ואחיות) של אנשים חולים, זוגותתכנון הריון. במקרה זה, הניתוח הכמותי של מספר הגנים של לוקוס SMA משחק תפקיד חשוב.
בדיקת DNA לפני לידה יכולה להפחית את הסיכוי ללדת תינוק עם מחלת ורדניג-הופמן. עם זאת, כדי להשיג חומר DNA עוברי, יש צורך להשתמש בשיטות פולשניות לאבחון טרום לידתי: בדיקת מי שפיר, ביופסיה כוריונית, קורדוקנטזה. אמיוטרופיה של ורדניג-הופמן, מאובחנת ברחם, היא אינדיקציה להפסקה מלאכותית של הריון.
טיפול באמיוטרופיה של Werdnig-Hoffmann
טיפול אטיופתוגני לא פותח. נכון לעכשיו, אמיוטרופיה של Werdnig-Hoffmann מטופלת על ידי שיפור המטבוליזם של פריפריה מערכת עצביםורקמת שריר כדי להאט את התקדמות הסימפטומים. בטיפול, שילובים של תרופות שונות קבוצות פרמקולוגיות: נוירומטבוליטים (תרופות המבוססות על הידרוליזט של מוח חזיר, ויטמינים גר. B, חומצה גמא-אמינו-בוטירית, פיראצטאם), מקלות על העברה עצבית-שרירית (גלנטמין, סנגוינארין, ניאוסטיגמין, איפידקרין), שיפור הטרופיזם המיופיברילי (חומצה גלוטמית, קו-אנזים Q10,, L-Carnitine). מתיונין), המשפרים את זרימת הדם (חומצה ניקוטינית, סקופולאמין). מומלץ לבצע תרגילי פיזיותרפיה ועיסוי ילדים.
ההתפתחות המודרנית של הטכנולוגיה אפשרה להקל במידת מה על חייהם של החולים וקרוביהם, הודות לשימוש בכיסאות גלגלים אוטומטיים ומאווררים ניידים. עזור לשפר את ניידות המטופל שיטות שונותתיקון אורטופדי. עם זאת, הסיכויים העיקריים בטיפול ב-SMA קשורים בפיתוח הגנטיקה ובחיפוש אחר הזדמנויות לתיקון סטיות גנטיות באמצעות שיטות. הנדסה גנטית.
תַחֲזִית
לאמיוטרופיה מולדת של Werdnig-Hoffman יש פרוגנוזה לא חיובית ביותר. כשהיא באה לידי ביטוי בימים הראשונים לחייו של ילד, מותו, ככלל, מתרחש לפני גיל 6 חודשים. בתחילת המרפאה לאחר 3 חודשי חיים מוות מתרחש בממוצע עד גיל שנתיים, לעיתים עד 7-8 שנים. צורת הילדות המוקדמת מאופיינת בהתקדמות איטית יותר, ילדים מתים בגיל 14-15 שנים.
תדירות מחלות
SMA היא אחת המחלות היתומות (נדירות) הנפוצות ביותר, הפוגעת ביילוד אחד ב-6000-10000.
גורם ל-SMA
SMA היא מחלה תורשתית הקשורה למוטציות בגן SMN1.
כדי שהמחלה תתבטא, שני ההורים חייבים להיות נשאים של המוטציה בגן זה. הגן הרצסיבי של SMA הוא אחד מכל 40 בערך. ההסתברות ללדת ילד חולה משני נשאים היא 25%, באותה הסתברות שלילד משני נשאים לא תהיה התמוטטות גנים. בעוד 50% מהמקרים הוא יהיה נשא של SMA, אך לא יחלה.
במקרים נדירים (פחות מ-2%), ילדים שנפגעו נולדים במשפחות שבהן רק הורה אחד הוא נשא. בהורה השני מתרחשת מוטציה בגן כאשר מוטלת ביצית או זרע.
מה נפגע ממוטציה
עקב גן פגום בגוף, ייצור חלבון SMN, חלבון ההישרדות של נוירונים מוטוריים, מופרע. ללא חלבון זה, נוירונים מוטוריים - תאי העצב של חוט השדרה האחראים על תיאום התנועות וטונוס השרירים - מתים, האות לשרירי הרגליים, הגב, ובחלקו הזרועות לא עובר.
ללא הטון הדרוש, השרירים מתנוונים בהדרגה. המחסור בשרירי הבטן והגב מוביל, בין היתר, לעיקול נרחב של עמוד השדרה, והם מובילים לבעיות נשימה, שכבר קיימות עקב שרירים חלשים.
המחלה יכולה להתבטא כבר מחודשי החיים הראשונים או בגיל מאוחר יותר.

מה קובע את חומרת המחלה
שני גנים אחראים לייצור חלבון SMN - SMN1 ו-SMN2.
יחד עם זאת, SMN1 הוא ה"לקוח" העיקרי של חלבון זה, ו-SMN2 הוא אחד נוסף, הוא מייצר חלבון בכמות שאינה מספקת לתפקוד תקין של הגוף. במקרים בהם SNM1 נעדר בגנום האנושי, SNM2 מתחיל לבצע פונקציות החלפה, אך לעולם לא יכול למלא את החסר במלואו.
ישנם עד שמונה עותקים של SMN2 בגנום. חומרת מצבו של המטופל תלויה במספר העותקים של SMN2 שיש לאדם. כגון מנגנון מורכבהמחלה מובילה לעובדה של-SMA יש כמה צורות, ומצבם של החולים שונה מאוד.
מהן הצורות של SMA?
ישנם 4 סוגים של SMA, הנבדלים זה מזה בחומרתם ובגיל שבו המחלה מופיעה לראשונה.
SMA I, מחלת ורדניג-הופמן.הצורה החמורה ביותר של המחלה מתרחשת אצל תינוקות בין גיל 0 ל-6 חודשים. ילדים עם צורה זו מלידה מתקשים לנשום, לינוק ולבלוע, וגם לא שולטים בתנועות המבוקרות הפשוטות ביותר - הם לא מחזיקים את הראש, הם לא יושבים בכוחות עצמם. בעבר חשבו שהרוב (80%) לא חי מעבר לגיל שנתיים. עכשיו, הודות לאסטרטגיות אוורור חדשות ו הזנה בצינורניתן להאריך את תוחלת החיים בעוד מספר חודשים.
SMA II, מחלת דובוביץ.הביטויים הראשונים של המחלה בגיל 7-18 חודשים. אדם עם SMA מסוג זה יכול לאכול ולשבת, אך אינו הולך באופן עצמאי. תוחלת החיים תלויה במידת הפגיעה בשרירים המספקים נשימה.
SMA III, מחלת קוגלברג-וולנדר.המחלה מתבטאת לראשונה לאחר שנה וחצי. חולים כאלה יכולים לעמוד (בכאב), אך לא ללכת. לתוחלת החיים של SMA סוג III, ככלל, אינו משפיע, אך פוגע מאוד באיכותו.
SMAIV, סוג זה נקרא גם "SMA למבוגרים",מאחר והמחלה מתבטאת בדרך כלל לאחר גיל 35 שנים.
תסמינים - חולשת שרירים, עקמת ורעד. בנוסף, מתפתחים התכווצויות מפרקים (הגבלה בתנועתיות במפרקים) והפרעות מטבוליות.
התקדמות המחלה אינה מהירה במיוחד, ראשית חולשת שרירים משפיעה על שרירי הרגליים, ולאחר מכן על הידיים. בדרך כלל בעיות בבליעה ו תפקוד נשימתיחולים לא.
רוב החולים עם SMA מסוג IV יכולים ללכת, ורק מעטים צריכים להיעזר בכסאות גלגלים.
SMA הקשור להפרעה בגן SMN, ב ספרות רפואיתנקראים פרוקסימליים - הם מהווים 95% מכלל האמיוטרופיות בעמוד השדרה. SMAs שאינם קשורים לגן SMN הם רבים למדי, אך הם נדירים. אלה כוללים, למשל, את מחלת קנדי. מחקר בשנות ה-90 הראה שמחלת קנדי אינה קשורה לפירוק של הגן SMN1, אלא למוטציות גנטיות אחרות המובילות לפגיעה בספיגה של חלבון SMN. המחלה מתבטאת באנשים מעל גיל 35. SMPA מאופיין בעיקר בחולשת גפיים.
סוג אחד של SMA שאינו קשור לגן SMN נקרא מחלת קנדי.העובדה שמחלה זו עדיין מכונה לפעמים SMA היא אנכרוניזם. בסוף שנות ה-60, מתי תיאור מפורטניוון זה, הוא נחשב לסוג של SMA, מכיוון שהוא משפיע על אותם עצבים ושרירים כמו ב שלושה סוגים SMA (אבל במידה הרבה פחות).
איך זה מטופל?
נכון להיום, אין תרופה ל-SMA.
התאגיד הבינלאומי "ביוג'ן" פיתח את התרופה "ספינרזה", ששיפרה משמעותית את מצבם של חולים אליהם שימשה במהלך הבדיקה. נכון להיום התרופה מאושרת לשימוש בארה"ב, באירופה העלות המשוערת של קורס שנתי, על פי חישובי החברה, תעמוד על כ-270 אלף יורו, ברוסיה התרופה אינה מאושרת. טיפול לכל החיים.
האם אפשר לעזור לאנשים עם SMA ואיך בדיוק?
עדיין לא ניתן לרפא את המחלה, אך ניתן להקל על מצבם של חולי SMA, כלומר, דרכים שונותלפצות על ביטויי המחלה.
בסוגים חמורים של SMA, יש לעזור לאנשים לנשום ולבלוע. לכן, טלפונים ניידים חיוניים עבורם. מאווררים, שואבי שיעול, שקיות Ambu.
ילדים עם SMA זקוקים גם לעזרה של מתנדבים שמסוגלים לפחות זמן קצרלהחליף הורים.
ילדים עם SMA עשויים להזדקק לעזרה בכל עת, אז אמהות ואבות תמיד ערים ולומדים את מיומנויות ההחייאה הדרושות למקרה שהילד יפסיק לנשום פתאום.
חולים פחות קשים זקוקים לתרופות להקלת הנשימה, מחוכים, כסאות גלגלים ומכשירים אחרים המקלים על התנועה והחיים של אנשים עם שרירים חלשים.
מחלה שנמשכת שנים רבות היא מתישה, ולכן חולים, בעיקר מבוגרים, זקוקים לא פעם לעזרת פסיכולוג.

הקרן פועלת ברחבי רוסיה. לעבודת הקרן שני כיוונים עיקריים - מתן סיוע לחולי SMA עצמם וליקיריהם, ועבודה על שינויים מערכתיים במצב עם SMA ברוסיה.
תוכלו לתמוך בפעילות הקרן על ידי תרומה בכל דרך הנוחה לכם. ניתן לסייע בתרומה חד פעמית או רגילה בעמוד מיוחד של הקרן או בשליחת SMS למספר הקצר 3443 עם המילה SMA ובהפרדה ברווח, סכום התרומה - למשל, CMA 300.
האם ניתן לקבל SMA מחיסונים?
באירופה ובארצות הברית לא אותר הקשר בין חיסונים לביטוי המחלה.
כדי להבין אם יש קשר בין SMA לחיסונים עשוי להסביר את ההבדל בין SMA לפוליו. פולומיאליטיס - הַדבָּקָהכאשר הגוף ניזוק מזיהום בתחילה ילד בריא. ילד עם SMA, שנולד עם גנום פגוע, עשוי להיראות בריא מבחוץ, אבל למעשה הוא כבר חולה, רק התסמינים של מחלתו מופיעים בהדרגה. בהקשר זה, SMA היא אותה מחלה "מושהית" כמו למשל, ניוון שרירים דושן או תסמונת רט, כאשר ילד המתפתח בהתאם לנורמה במשך זמן מה מאבד מיומנויות שנרכשו בעבר והופך לנכה.
רוב הביטויים של SMA קשורים לפיתוח המיומנויות המוטוריות הראשונות. הביטויים הראשונים של המחלה חופפים בזמן למספר חיסונים הקשורים לגיל. כתוצאה מכך, אדם וקרוביו עשויים לטעון כי הוא "חלה מהחיסון", אך למעשה הוא רק הראה סימני מחלה שכבר היו לו.
כיצד נקבע שלילד יש SMA ולא מחלה אחרת?
למרות העובדה ש-SMA תוארה לראשונה על ידי הנוירולוג האוסטרי גווידו ורדניג והנוירולוג הגרמני יוהאן הופמן בתחילת שנות ה-90, אופי המחלה הובן במלואו רק בסוף המאה ה-20. הגן SMN1 התגלה בשנת 1995. כדי לאשר את האבחנה של SMA, יש צורך בבדיקה גנטית.
ברוסיה, בדיקות גנטיות מתאימות הפכו לזמינות בתחילת שנות ה-2000. אפשר לעשות בדיקה גנטית ל-SMA לפי ביטוח בריאות חובה, אבל בפועל, לא יותר מדי רופאים מכירים את האבחנה הנדירה הזו ומפנים מטופלים למחקר מתאים. העלות של בדיקה כזו מעבדות מסחריותמוסקבה - כ -6 אלף רובל.
היעדר אבחון ספציפי הוביל גם לבלבול באבחונים. רוב החולים ב-SMA ברוסיה לא זוהו, רבים מאלה שזוהו אובחנו עם מחלת ורדניג-הופמן, אם כי לא כולם (במיוחד מבוגרים) אכן חולים בסוג זה של מחלה.
כמה חולים עם SMA יש ברוסיה?

התרופה "Spinraza", אשר משפרת באופן משמעותי את מצב החולים. תמונה מאת healthbeat.spectrumhealth.org
בהתחשב בתדירות המחלה, מספר החולים ב-SMA ברוסיה צריך להיות בין שבעה לעשרים וארבעה אלף איש. נכון להיום, ישנם כ-400 איש בפנקס המטופלים של קרן משפחות SMA.
מי ברוסיה עוזר לאנשים עם SMA ולמשפחותיהם
קרן צדקה ורה, בית הוספיס לילדים עם מגדלור, קרן צדקה פליאטיבית לילדים, קרן צדקה משפחות SMA, שירות פליאטיבי לילדים רחמים.
משנת 2014 מתפתח במוסקבה פרויקט משותף של השירות "רחמים" וקרן "משפחות SMA" "מרפאות SMA. במפגשים המתקיימים אחת לחודש יכולים המטופלים לקבל ייעוץ מרופא ריאות, אורטופד, פיזיותרפיסט ו פְּסִיכוֹלוֹג. IN לָאַחֲרוֹנָהחלק מהמפגשים מתמקדים גם בצרכים של מטופלים מבוגרים.
אנשים מפורסמים עם SMA

סימון שפינוגליו האיטלקייהנולד עם מחלה תורשתיתניוון שרירי עמוד השדרה סוג 2. היא לא מסוגלת ללכת מאז הלידה ומתנועעת רק בעזרת כסא גלגלים חשמלי. אבל חייה מלאים ועשירים; שום דבר לא יכול לעצור את הרצון שלה לחיות.
סימון עובדת עבור קו חם» של ה-Italian Famiglies of SMA Association ומסייע לילדים ומבוגרים עם SMA ומחלות עצב-שריר אחרות.
סימון גם הקליטה כמה שירים פופולריים בקהילת SMA האיטלקית - על החופש לעשות מה שאתה רוצה, למרות המחלה.
הזמרת הרוסייה יוליה סמוילובהנולדה באוכטה (רפובליקה קומי) בגיל עשר הופיעה בקונצרט צדקה, ולאחר מכן הוזמנה לשיר בארמון החלוצים המקומי. בגיל חמש עשרה החלה ללמוד בבית התרבות של העיר.
ב-2008 היא הרכיבה משלה קבוצה מוזיקלית(פורק ב-2010). בשנת 2013 היא השתתפה בתחרות פקטור A בערוץ הטלוויזיה רוסייה. היא זכתה במקום השני וקיבלה את הפרס האישי של אללה פוגצ'בה "כוכב הזהב של אללה". בשנת 2017, עקב אי קבלתה של רוסיה ל תוכנית תחרותיתלא הצליח להשתתף באירוויזיון. נע בכיסא גלגלים.
מתכנת מוולדימיר ולרי ספירידונוב. הוא סיים את בית הספר עם מדליית זהב, ואז הגן על תואר ההנדסה שלו. בשנת 2015, ולרי תכננה להיות משתתפת בניסוי של המנתח האיטלקי סרג'יו קנוברו להשתלת ראש אנושי (הניסוי בוטל).
כיום ולרי הוא חבר בלשכה הציבורית בעיר של ולדימיר, מומחה לעניין סביבה נגישה, וכן היוצר של קהילה משלו "תשוקה לחיים", המדברת על יצירת סביבה נגישה ופרויקטים רפואיים מבטיחים. ולרי משתתפת בתוכניות טלוויזיה רבות בטלוויזיה הרוסית והזרה.
מהי אמיוטרופיה של עמוד השדרה ורדניג-הופמן?
אמיוטרופיה של עמוד השדרה של ורדניג-הופמן.המחלה תוארה על ידי J. Werdnig בשנת 1891 ו-J. Hoffmann בשנת 1893. השכיחות היא 1 ל-100,000 אוכלוסייה, 7 ל-100,000 יילודים.
מה מעורר אמיוטרופיה של עמוד השדרה ורדניג-הופמן
זה עובר בתורשה בצורה אוטוזומלית רצסיבית.
פתוגנזה (מה קורה?) במהלך עמוד השדרה Werdnig-Hoffmann Amyotrophy
תת התפתחות של תאים של הקרניים הקדמיות של חוט השדרה, דה-מיילינציה של השורשים הקדמיים נמצאים. לעתים קרובות יש שינויים דומים בגרעינים המוטוריים ובשורשים V, VI, VII, IX, X, XI ו- XII עצבים גולגולתיים. בשרירי השלד, שינויים נוירוגניים מאופיינים ב"ניוון צרור", חילופין של צרורות סיבי שריר מנוונים ושמורים, כמו גם הפרעות אופייניות למיאופתיות ראשוניות (היאלינוזה, היפרטרופיה של סיבי שריר בודדים, היפרפלזיה רקמת חיבור).
תסמינים של אמיוטרופיה של עמוד השדרה Werdnig-Hoffmann
ישנן שלוש צורות של המחלה: מולדת, ילדות מוקדמת ומאוחרת, שונות בזמן הביטוי של הראשון. תסמינים קלינייםוקצב התהליך האמיוטרופי.
בְּ צורה מולדת מהימים הראשונים של החיים בילדים, היפוטוניה שרירית כללית והיפוטרופיה של השרירים, מתבטאים ירידה או היעדר רפלקסים בגידים. הפרעות בולברי נקבעות מוקדם, המתבטאות במציצה איטית, בכי חלש, פרפור הלשון וירידה ברפלקס הלוע. המחלה משולבת עם עיוותים אוסטיאוארטיקולריים: עקמת, חזה בצורת משפך או "עוף", התכווצויות מפרקים. התפתחות תפקודים סטטיים ותנועתיים מואטת בחדות. רק מספר מצומצם של ילדים, באיחור גדול, מפתח את היכולת להחזיק את הראש ולהתיישב בכוחות עצמם. עם זאת, מיומנויות מוטוריות נרכשות נסוגות במהירות. ילדים רבים עם צורה מולדת של המחלה הפחיתו את האינטליגנציה. נצפה לעתים קרובות מומים מולדיםהתפתחות: הידרוצפלוס מולד, קריפטורכידיזם, המנגיומה, דיספלזיה של מפרק הירך, כף הרגל וכו'.
זְרִימָה. מהלך המחלה מתקדם במהירות. מוותמתרחש לפני גיל 9. אחד מגורמי המוות העיקריים הוא הפרעות סומטיות קשות (אי ספיקת לב וכלי דם ונשימה), הנגרמות מחולשה של שרירי החזה וירידה בהשתתפותו בפיזיולוגיה של הנשימה.
בְּ צורת ילדות מוקדמת הסימנים הראשונים של המחלה מופיעים, ככלל, במחצית השנייה של החיים. ההתפתחות המוטורית במהלך החודשים הראשונים משביעת רצון. ילדים במועד מתחילים להחזיק את ראשם, לשבת, לפעמים לעמוד. המחלה מתפתחת בצורה תת-חריפה, לעתים קרובות לאחר זיהום, שיכרון מזון. פרזיס רפוי הוא בתחילה מקומי ברגליים, ואז מתפשט במהירות לשרירי תא המטען והזרועות. ניוון שרירים מפושט משולבת עם פקקולציות, פרפורי הלשון, רעד עדין של האצבעות, התכווצויות גידים. טונוס שרירים, רפלקסים של גידים ופריוסטאליים מופחתים. בשלבים המאוחרים יותר, יש תת לחץ דם כללי, תופעות של שיתוק בולברי.
זְרִימָה. ממאיר, אם כי מתון יותר מהצורה המולדת. תוצאה קטלנית מתרחשת בגיל 14-15 שנים.
בְּ צורה מאוחרת הסימנים הראשונים של המחלה מופיעים בגיל 1.5-2.5 שנים. עד גיל זה, היווצרות של תפקודים סטטיים ותנועתיים הושלמה לחלוטין אצל ילדים. רוב הילדים הולכים ורצים בכוחות עצמם. המחלה מתחילה באופן בלתי מורגש. התנועות הופכות למסורבלות, לא בטוחות. ילדים לעתים קרובות מועדים ונופלים. ההליכה משתנה: הם הולכים עם ברכיים כפופות (הליכת "בובת השעון"). פרזיס רפוי מתמקם בתחילה בקבוצות השרירים הפרוקסימליות של הגפיים התחתונות, ולאחר מכן נע באיטיות יחסית לקבוצות השרירים הפרוקסימליות של הגפיים העליונות, שרירי תא המטען; ניוון שרירים הוא בדרך כלל עדין עקב שכבת שומן תת עורית מפותחת היטב. פאסיקולציות אופייניות, רעד עדין של האצבעות, תסמינים בולבריים - פרפור וניוון של הלשון, ירידה ברפלקסים של הלוע והפלטין. רפלקסים של גידים ופריוסטאליים נמוגים כבר שלבים מוקדמיםמחלה. עיוותים אוסטיאוארטיקולריים מתפתחים במקביל למחלה הבסיסית. העיוות הבולט ביותר של החזה.
זְרִימָה. ממאיר, אבל רך יותר משתי הצורות הראשונות. הפרה של היכולת ללכת באופן עצמאי מתרחשת בגיל 10-12 שנים. חולים חיים עד 20-30 שנים.
אבחון של אמיוטרופיה של עמוד השדרה של ורדניג-הופמן
האבחנה מבוססת על ניתוח גנאלוגי (תורשה אוטוזומלית רצסיבית), מאפיינים קליניים (התפרצות מוקדמת, ניוון מפוזר עם לוקליזציה דומיננטית בקבוצות השרירים הפרוקסימליות, יתר לחץ דם כללי בשרירים, פקקולציות ופרפורים של הלשון, היעדר פסאודו-היפרטרופיה, פרוגרדיאנט וברוב המקרים מהלך ממאיר וכו'), תוצאות של אלקטרומיוגרפיה גלובלית (עורית) ומחקרים מורפולוגיים של שרירי השלד, החושפים את אופי הדנרבציה של השינויים.
יש להבדיל בין צורות מולדות ומוקדמות בעיקר ממחלות הכלולות בקבוצות של תסמונות עם יתר לחץ דם מולד (תסמונת "ילד איטי): אופנהיים אמיאטוניה, צורה מולדת שפירה של ניוון שרירים, צורה אטונית של שיתוק מוחין תינוקות, חומרים תורשתיים למחלות מטבוליות, תסמונות כרומוזומליות וכו' יש להבדיל בין הצורה המאוחרת לאמיוטרופיה של עמוד השדרה של קוגלברג-וולנדר, ניוון שרירים פרוגרסיבי של דושן, ארבה-רוט וכו'.
טיפול באמיוטרופיה של עמוד השדרה Werdnig-Hoffmann
עם אמיוטרופיה של עמוד השדרה של Werdnig-Hoffmann, טיפול בפעילות גופנית, עיסוי, תרופות המשפרות את הטרופיזם של רקמת העצבים נקבעות - cerebrolysin, aminalon (gammalon), pyriditol (encephabol).
לאילו רופאים עליך לפנות אם יש לך אמיוטרופיה של עמוד השדרה Werdnig-Hoffmann
נוירולוג
מבצעים ומבצעים מיוחדים
חדשות רפואיות
14.11.2019
מומחים מסכימים כי יש צורך למשוך את תשומת הלב הציבורית לבעיות מחלת לב וכלי דם. חלקם נדירים, מתקדמים וקשים לאבחון. אלה כוללים, למשל, קרדיומיופתיה עמילואידית טרנסטירטין.
14.10.2019
ב-12, 13 ו-14 באוקטובר, רוסיה מארחת קמפיין חברתי רחב היקף לבדיקת קרישת דם בחינם - "יום INR". הפעולה מתוזמן ל יום העולםהמאבק נגד פקקת. 04/05/2019
השכיחות של שעלת בפדרציה הרוסית בשנת 2018 (לעומת 2017) כמעט הוכפלה1, כולל בילדים מתחת לגיל 14. מספר כוללהמקרים הרשומים של שעלת בינואר-דצמבר עלו מ-5,415 מקרים בשנת 2017 ל-10,421 מקרים עבור אותה תקופה בשנת 2018. השכיחות של שעלת עולה בהתמדה מאז 2008...
מאמרים רפואיים
כמעט 5% מכולם גידולים ממאיריםמהווים סרקומות. הם מאופיינים באגרסיביות גבוהה, התפשטות המטוגנית מהירה ונטייה להישנות לאחר הטיפול. חלק מהסרקומות מתפתחות במשך שנים מבלי להראות דבר...
וירוסים לא רק מרחפים באוויר, אלא יכולים גם לעלות על מעקות, מושבים ומשטחים אחרים, תוך שמירה על פעילותם. לכן, בעת נסיעה או במקומות ציבוריים, רצוי לא רק לא לכלול תקשורת עם אנשים אחרים, אלא גם להימנע ...
לַחֲזוֹר ראיה טובהולנצח להיפרד ממשקפיים ועדשות מגע - חלומם של אנשים רבים. כעת ניתן להפוך אותו למציאות במהירות ובבטחה. הזדמנויות חדשות תיקון לייזרהראייה נפתחת בטכניקת Femto-LASIK ללא מגע לחלוטין.
תכשירים קוסמטיים שנועדו לטפל בעור ובשיער שלנו אולי לא באמת בטוחים כמו שאנחנו חושבים.
- חולשה בשרירים: סרבול בזמן תנועות, הפרעה בהליכה (שלב עם כיפוף רגליים בברכיים).
- ירידה בטונוס השרירים.
- הפחתת נפח הגפיים עקב חוסר התפתחות של השרירים.
- רעד (עוויתות קטנות, רעידות) של הגפיים.
- ביטוי מופחת.
- הפרעת בליעה – חנק בעת בליעה.
- אי ספיקת נשימה וכתוצאה מכך ברונכיטיס תכופה, דלקת ריאות (דלקת רקמת הריאות). זה מתרחש עקב תקלה של שרירי הנשימה, בעוד שקיפאון דם מתפתח בריאות. זהו מצב הנוטה להתפתחות דלקת.
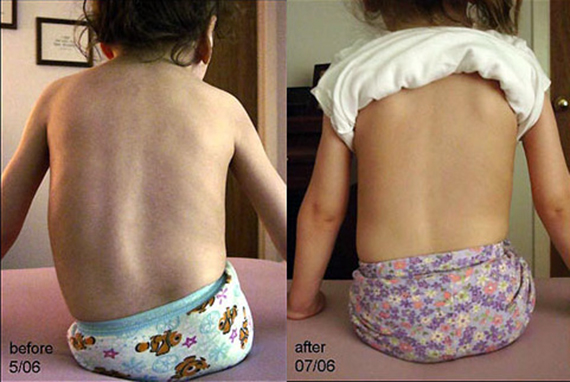
- עיוותים של המנגנון האוסטיאוארטיקולרי: חזה ("עוף", חזה בצורת משפך), עמוד השדרה (עקמת - עקמומיות של עמוד השדרה).
- ירידה בלתי הפיכה בניידות במפרקי הגפיים (חוזים).
- האטה בהתפתחות הפיזית של הילד.
טפסים
- מולד -תסמינים נצפים מיד לאחר הלידה:
- הילד רדום;
- לוקח את השד בצורה גרועה;
- הבכי חלש;
- מגיב באיטיות לסביבה;
- צורה זו משולבת לעתים קרובות עם סימנים של התפתחות שלד לקויה (עקמת - עקמומיות של עמוד השדרה, עיוות של החזה).
- מוקדם של ילדים:הילד מתפתח כרגיל עד שישה חודשים, ואז (לעתים קרובות לאחר מכן זיהום במעיים) מפתחים שיתוק תחילה של הרגליים, אחר כך של הידיים, טונוס השרירים יורד. ילדים לא מחזיקים את הראש, לא יושבים, לא מתהפכים.
- מאוחר:הסימנים הראשונים מופיעים לאחר 1-3 שנים, לעתים קרובות הופעת הסימנים הראשונים לא מורגשת. הילד מתפתח כרגיל, ואז ההליכה מתחילה להישבר, התנועות הופכות למסורבלות, ואז מצטרפת ירידה כללית בטונוס השרירים.
גורם ל
- המחלה היא תורשתית, כלומר, היא מועברת מהורים לילדים.
אבחון
אם מופיעים התסמינים לעיל, צור קשר עם או.
הרופא יבצע בדיקה מלאה של הילד, יערוך בדיקה נוירולוגית וידון עם ההורים בסדר ובעיתוי התפתחות סימני המחלה.
חשוב לאסוף נתונים על נוכחות אפשרית של סימני המחלה אצל מישהו מהמשפחה, מה שעשוי לעורר את הרופא לחשוב על האופי התורשתי של המחלה.
כדי לאשר את האבחנה, הרופא רושם:
- ENMG (אלקטרונורומיוגרפיה): חיישנים על העור מתעדים מעבר של דחף עצבי. השיטה מאפשרת לקבוע לאילו הפרעות מוטוריות קשורות;
- ביופסיית שריר: חלק קטן מהשריר נלקח לבדיקה עם מחט;
- MRI (הדמיית תהודה מגנטית) של הראש: כדי לא לכלול פתולוגיה אחרת (השיטה מאפשרת לך ללמוד את המבנה של כל חלק בגוף בשכבות);
- ניתוח גנטי (מחקר של מבנה הכרומוזומים, חיפוש מוטציות גנטיות) היא שיטה המאששת את האבחנה.
טיפול באמיוטרופיה של עמוד השדרה של ילדים ורדניג-הופמן
אין כיום שיטות או תרופות המשפיעות על סיבת המחלה.
שיטות משמשות לשיפור התזונה של מערכת העצבים:
- טיפול בוויטמין;
- nootropics (תרופות המשפרות את תזונת המוח);
- לְעַסוֹת;
- פִיסִיוֹתֶרָפִּיָה.
סיבוכים והשלכות
- אִי תְזוּזָה.
- הפסקת נשימה.
- הסיכון למוות.
זְרִימָה צורה מוקדמתרך יותר: המוות מתרחש בגיל 14-15 שנים.
עם צורה מאוחרת, ילדים מאבדים את היכולת ללכת עד גיל 10-12, חיים עד 20-30 שנים.
מניעת אמיוטרופיה של עמוד השדרה של ילדים ורדניג-הופמן
- בשל אופייה התורשתי של המחלה, מניעה אינה אפשרית.