Spinale Amyotrophie (spinale Muskelatrophie oder SMA) ist eine unheilbare, fast immer vererbte Krankheit, die durch eine Genmutation auf Chromosom 5 verursacht wird.
Eine Mutation des SMA-Gens führt vor allem zu einem Mangel an Protein, das für den Aufbau von Protein-RNA-Strukturen notwendig ist, und zu einer unzureichenden Entwicklung der quergestreiften Muskulatur untere Gliedmaßen, und auch Halswirbelsäule und Köpfe.
Die Krankheit kann sich ab dem Zeitpunkt der Geburt und auch dann, wenn sich der Fötus noch im Mutterleib befindet, und in jedem Lebensabschnitt manifestieren. Bei Neugeborenen führt die spinale Amyotrophie am häufigsten zum frühen Tod, kann aber in einigen Fällen, vor allem im Alter, leichte Formen annehmen. Lassen Sie uns die Merkmale dieser Pathologie genauer betrachten.
Spinale Amyotrophie – alles über die Krankheit
Geschichte und Statistik
SMA reicht aus seltene Krankheit, offen Deutscher Arzt Werdnig im Jahr 1891. Einer von 6-10.000 Menschen erkrankt daran, aber jeder 50. Mensch ist Träger des rezessiven SMA-Gens.
Im Jahr 1898 stellten Werdnig und ein anderer Wissenschaftler, Hoffmann, fest, dass die Ursache von SMA eine degenerative Läsion und eine unzureichende Anzahl von Motoneuronen in den Vorderhörnern ist Rückenmark— SMN (Überlebensmotorneuronen).

Bereits im 20. Jahrhundert (1956) entdeckten die anderen Wissenschaftler Kugelberg und Welander eine weniger bösartige Form der SMA mit milderen Manifestationen, die Jugendliche und Erwachsene betrifft.
Art der Vererbung der spinalen Amyotrophie
Die Krankheit kann durch eine der folgenden Arten vererbt werden:
- autosomal-dominant;
- autosomal-rezessiv;
- X-chromosomal dominant;
- X-chromosomal rezessiv.
In dieser Hinsicht sind viele Dinge klassifiziert verschiedene Formen SMA.
Die Kindheitsform von SMA wird autosomal-rezessiv vererbt: Wenn beide Elternteile Träger sind, ist ein Viertel ihrer Nachkommen betroffen.
Der autosomal-dominante Erbgang der SMA führt mit einer Wahrscheinlichkeit von 50 % zur Manifestation der Erkrankung bei Kindern, auch wenn nur ein Elternteil erkrankt ist.
Arten der spinalen Amyotrophie
Die spinale Muskelatrophie wird in vier Formen unterteilt:
- Infantil (I) – Spinale Muskelatrophie nach Werdnig-Hoffmann: diagnostiziert von der Geburt bis zum sechsten Monat.
- Mittelstufe (II) – Dubowitz-Krankheit: von sieben Monaten bis eineinhalb Jahren.
- Jugend (III) – geb. Kugelberg-Welander: nach eineinhalb Jahren.
- Erwachsener (IV): nach 35 Jahren.
Symptome einer spinalen Muskelatrophie
Häufige Symptome von SMA:
- Schädigung der proximalen (mittleren) Muskeln und Faszien;
- weitgehende Erhaltung der Sensibilität klinische Fälle;
- geistige Verzögerungen und geistige Entwicklung mit spinaler Muskelamyotrophie sind äußerst selten;
- Bei einigen Arten ist eine Atrophie nicht nur der Muskeln der Gliedmaßen, sondern auch der Atem-, Kau- und Schluckmuskulatur möglich.

SMA-Schweregrad
- Als schwerster und ungünstigster Typ gilt die erste Form der SMA (infantil) – die Werdnig-Hoffman-Amyotrophie der Wirbelsäule, bei der Kinder nicht in der Lage sind, aktive Bewegungen auszuführen, den Kopf hochzuhalten oder selbstständig zu sitzen. Das Füttern ist für das Baby sehr schwierig, da es für ihn schwierig ist, Milch zu saugen und zu schlucken.
- Die Dubowitz-Krankheit (II. Zwischenform der SMA) ist weniger bösartig: Bei ihr können Kinder zwar sitzen, den Kopf hochhalten und essen, aber noch nicht laufen.
- Die juvenile Form ist am wenigsten schwerwiegend: Trotz Muskelschwäche kann das Kind laufen lernen, die Krankheit schreitet jedoch langsam voran und kann zu einer frühen Behinderung führen.
- Vierte Erwachsenenform Aufgrund einer Schwäche der proximalen Muskulatur kann es bei SMA zu Bewegungsunfähigkeit und Reflexverlust kommen, die Prognose für die Lebenserwartung bleibt jedoch günstig.

Andere Formen der spinalen Muskelatrophie
Zusätzlich zu Mutationen in Genen, Schaden verursachen proximale Muskeln, es gibt ähnliche Pathologien mit verschiedene Typen Vererbung, die zur Atrophie der Muskeln und Faszien der distalen (Endabschnitte) führt.
Die Liste davon ist ziemlich umfangreich. Fassen wir die Krankheiten in einer kleinen Tabelle zusammen:
| Name von SMA | Vererbungstyp | Merkmale und Symptome |
| SMAX1 | X-chromosomal rezessiv | Es wird hauptsächlich bei älteren Menschen beobachtet, betrifft die Bulbarnerven des Schädels und verursacht eine absteigende Lähmung. |
| SMAХ2 | X - Kupplung. rezessiv | Angeboren aggressive Form, was bis zu 3 Monate lang zum Tod führt. Verursacht Schwäche, Areflexie, Kontrakturen und Brüche. |
| SMAX3 | X - Kupplung. rezessiv | Betroffen sind vor allem Jungen. Atrophie aller distalen Muskeln. Langsames Fortschreiten der Symptome |
| Distales DSMA1 | autosomal-rezessiv | Angeboren, betrifft vor allem die Hände, schwere Atemwegsbeschwerden sind möglich |
| Distale Formen DSMA2 - DSMA5 | autosomal-rezessiv | Alle vier Formen zeichnen sich durch ein langsames Fortschreiten aus; DSMA5 wird bei jungen Menschen diagnostiziert. |
| Distale MCA von zwei Typen: VA und VB (DSMAVA und DSMAVB) | autosomal-dominant | Die oberen Gliedmaßen sind überwiegend verkümmert. |
| DSMA-Typ 2D | autosomal-rezessiv | Jugend und Erwachsenenkrankheit mit langsamer Entwicklung: Sowohl die proximale als auch die distale Muskulatur sind betroffen, zuerst in den Beinen, dann in den Armen. |
| DSMA Typ 7A | autosomal-dominant | Eine sehr seltene erwachsene Form mit Schädigung der Stimmbänder. |
| DSMA Typ 2A | autosomal-dominant | Art der Charcot-Krankheit (allelischer Typ) |
| Juvenile SMA (Typ HMN1) | autosomal-dominant | Tritt in der Jugend auf |
| Angeborene Amyotrophie der Wirbelsäule | autosomal-dominant | Innervationsstörung und Atrophie der Hüft-, Fuß- und Kniemuskulatur mit Kontraktur und Deformation; manchmal sind die Stimmbänder betroffen. |
| Finkels SMA | autosomal-dominant | Sie beginnt hauptsächlich im Alter von 35 bis 37 Jahren, es wurden jedoch auch Fälle der Krankheit im Kindesalter registriert. Es entwickelt sich langsam zuerst in den Beinen und dann in den Armen. Aktivität und Reflexe sind vermindert und es kommt zu unwillkürlichem Zittern (Faszikulation). |
| SMA Witza | autosomal-dominant | Betroffen sind erwachsene Prox. und distale Muskeln. |
| SMA (LED1-Typ) | autosomal-dominant | Atrophie der unteren Extremitäten bei Neugeborenen. |
| SMA-Typ PME | autosomal-rezessiv | Atrophie der distalen Muskulatur mit gestörter Innervation und epileptischen Anfällen |
| SMA mit angeborenen Knochenbrüchen | autosomal-rezessiv | Schwere Symptome, wie Krankheit. Werdnig-Hoffman, verschlimmert durch Frakturen. |
| SMA mit Hypoplasie | autosomal-dominant | Angeborene Hirnanomalie mit zerebralen Symptomen, Mikrozephalie und Entwicklungsverzögerung. |
| SMA juveniler asymmetrischer Typ | -------------- | Junge indische Männer leiden darunter |

In dieser Tabelle sollten Sie auf die letzten beiden Arten der spinalen Amyotrophie achten:
- SMA mit Hypoplasie geht mit Abweichungen in der geistigen und geistigen Entwicklung einher, die für andere Krankheitsarten nicht typisch sind.
- Asymmetrische juvenile (indische) Amyotrophie wird nicht vererbt. In diesem Fall kann sich die Krankheit nach zwei bis fünf Jahren schleppenden Verlaufs stabilisieren. Faszikulationssymptome sind bei dieser speziellen Form selten.
Behandlung der spinalen Amyotrophie
Es ist grundsätzlich unmöglich, diese Art von Krankheit zu heilen, wie jede erbliche Pathologie, die das Rückenmark oder das Gehirn betrifft. Die Wirksamkeit vieler Medikamente, die heute zur Behandlung von Amyotrophie eingesetzt werden, ist nicht nachgewiesen. Der Kern der Behandlung besteht darin, das Protein zu erhöhen, das an der Bildung von SMN-Motoneuronen beteiligt ist.
Behandlungsmöglichkeiten für SMA
Daher werden hauptsächlich folgende Medikamente verwendet:
- Valproinsäure;
- Natriumhydroxybutyrat;
- Nusinersen (ein neues Medikament, das 2016 in den USA zur Behandlung von SMA eingeführt wurde).
Unterstützende Behandlung (Physiotherapie, Massage, Krankengymnastik), speziell Protein-Diät, was berücksichtigt mögliche Kontraindikationen. Patienten, die nicht in der Lage sind, sich selbstständig zu bewegen, benötigen soziale Betreuung.
Hierbei handelt es sich um die bösartigste spinale Muskelatrophie, die sich von Geburt an oder in den ersten 1–1,5 Lebensjahren eines Kindes entwickelt. Sie ist gekennzeichnet durch eine zunehmende diffuse Muskelatrophie, begleitet von einer schlaffen Parese, die bis zur vollständigen Plegie fortschreitet. In der Regel geht die Werdnig-Hoffmann-Amyotrophie mit Knochendeformitäten und angeborenen Entwicklungsanomalien einher. Die diagnostische Grundlage sind Anamnese, neurologische Untersuchung, elektrophysiologische und tomographische Untersuchungen, DNA-Analyse und Untersuchung der morphologischen Struktur Muskelgewebe. Die Behandlung ist schwach wirksam und zielt auf die Optimierung des Trophismus des Nerven- und Muskelgewebes ab.
ICD-10
G12.0 Pädiatrische spinale Muskelatrophie, Typ I [Werdnig-Hoffmann]

allgemeine Informationen
Die Werdnig-Hoffmann-Amyotrophie ist die schwerste Variante aller spinalen Muskelatrophien (SMA). Die Prävalenz beträgt 1 Fall pro 6.000 bis 10.000 Neugeborene. Jeder 50. Mensch ist Träger des veränderten Gens, das die Krankheit verursacht. Dank der autosomal-rezessiven Vererbung manifestiert sich die Pathologie bei einem Kind jedoch nur dann, wenn die entsprechende genetische Aberration sowohl bei der Mutter als auch beim Vater vorliegt. Die Wahrscheinlichkeit, in einer solchen Situation ein Kind mit einer Pathologie zu bekommen, beträgt 25 %.
Die Krankheit hat verschiedene Formen: angeboren, mittelschwer (frühe Kindheit) und spät. Eine Reihe von Spezialisten identifizieren die letztere Form als eigenständige Nosologie – die Kugelberg-Welander-Amyotrophie. Der Mangel an etiotroper und pathogenetischer Behandlung und der frühe Tod machen die Behandlung von Patienten mit Werdnig-Hoffmann-Krankheit zu einer der schwierigsten komplexe Aufgaben Herausforderungen der modernen Neurologie und Pädiatrie.
Ursachen
Die Werdnig-Hoffmann-Amyotrophie ist eine erbliche Pathologie, die durch einen Zusammenbruch des genetischen Apparats auf der Ebene des 5q13-Locus des 5. Chromosoms verursacht wird. Das Gen, in dem Mutationen auftreten, wird als Survival Moto Neuron Gene (SMN) bezeichnet – das Gen, das für das Überleben von Motoneuronen verantwortlich ist. 95 % der Patienten mit Werdnig-Hoffmann-Krankheit weisen eine Deletion der telomeren Kopie dieses Gens auf. Der Schweregrad der SMA korreliert direkt mit der Länge der Deletionsstelle und dem damit einhergehenden Vorhandensein von Veränderungen (Rekombination) in den Genen H4F5, NAIP und GTF2H2.
Das Ergebnis der Aberration des SMN-Gens ist die Unterentwicklung der Motoneuronen des Rückenmarks, die in seinen Vorderhörnern lokalisiert sind. Die Folge ist eine unzureichende Innervation der Muskulatur, die zu einer starken Atrophie mit Verlust der Muskelkraft und einem fortschreitenden Rückgang der Fähigkeit zu aktiven motorischen Handlungen führt. Die Hauptgefahr ist Muskelschwäche Brust, ohne deren Beteiligung Bewegungen, die die Atemfunktion sicherstellen, unmöglich sind. Gleichzeitig bleibt die Sinnessphäre während der gesamten Krankheitsdauer intakt.
Symptome einer Amyotrophie
Angeborene Form(SMA I) manifestiert sich klinisch vor dem 6. Lebensmonat. In der Gebärmutter kann es sich durch eine träge Bewegung des Fötus äußern. Oft wird bereits in den ersten Lebenstagen eine Muskelhypotonie festgestellt, die mit dem Aussterben tiefer Reflexe einhergeht. Kinder weinen schwach, saugen schlecht und können ihren Kopf nicht hochhalten. In einigen Fällen (bei späterem Einsetzen der Symptome) lernt das Kind, den Kopf hochzuhalten und sogar zu sitzen, aber mit fortschreitender Krankheit verschwinden diese Fähigkeiten schnell. Charakteristisch sind frühe Bulbusstörungen, verminderter Rachenreflex und faszikuläres Zucken der Zunge.
Diese Werdnig-Hoffmann-Amyotrophie geht mit Oligophrenie und Störungen der Ausbildung des Knochen- und Gelenkapparates einher: Brustdeformitäten (Trichter- und Kielbrust), Wirbelsäulenverkrümmung (Skoliose), Gelenkkontrakturen. Viele Patienten haben andere angeborene Anomalien: Hämangiome, Hydrozephalus, Klumpfuß, Dysplasie Hüftgelenke, Kryptorchismus usw.
Der Verlauf der SMA I ist am bösartigsten, mit rasch zunehmender Immobilität und Parese der Atemmuskulatur. Letzteres verursacht die Entwicklung und das Fortschreiten von Atemversagen, das die Haupttodesursache darstellt. Aufgrund der Schluckbeschwerden kann es passieren, dass Lebensmittel in den Mund geworfen werden Fluglinien mit der Entwicklung einer Aspirationspneumonie, die eine tödliche Komplikation einer spinalen Amyotrophie sein kann.
Frühkindliche Form(SMA II) debütiert im Alter von 6 Monaten. Zu diesem Zeitpunkt haben Kinder eine dem Alter entsprechende zufriedenstellende körperliche und neuropsychische Entwicklung erreicht; sie erwerben die Fähigkeiten, den Kopf zu halten, sich umzudrehen, sich hinzusetzen und zu stehen. Doch in den allermeisten klinischen Fällen haben Kinder keine Zeit, laufen zu lernen. Typischerweise manifestiert sich diese Werdnig-Hoffmann-Amyotrophie, nachdem ein Kind eine lebensmittelbedingte toxische Infektion oder eine andere akute Infektionskrankheit erlitten hat.
In der Anfangsphase kommt es zu peripheren Paresen in den unteren Extremitäten. Dann breiten sie sich schnell auf die oberen Gliedmaßen und die Rumpfmuskulatur aus. Es entwickelt sich eine diffuse Muskelhypotonie und die Tiefenreflexe lassen nach. Es werden Sehnenkontrakturen, Fingerzittern und unwillkürliche Muskelkontraktionen (Faszikulationen) der Zunge beobachtet. In späteren Stadien treten Bulbärsymptome und fortschreitendes Atemversagen auf. Der Verlauf ist langsamer als bei der angeborenen Form der Werdnig-Hoffmann-Krankheit. Patienten können bis zu 15 Jahre alt werden.
Kugelberg-Welander-Amyotrophie(SMA III) – die harmloseste spinale Amyotrophie Kindheit. Manifestiert sich nach 2 Jahren, in einigen Fällen im Zeitraum von 15 bis 30 Jahren. Es gibt keine geistige Behinderung, lange Zeit Patienten können sich selbstständig bewegen. Einige von ihnen leben bis ins hohe Alter, ohne die Fähigkeit zur Selbstfürsorge zu verlieren.
Diagnose
Aus diagnostischer Sicht sind für einen Kinderneurologen das Alter des Auftretens der ersten Symptome und die Dynamik ihrer Entwicklung sowie Daten zum neurologischen Status (vor allem das Vorliegen) wichtig motorische Störungen peripherer Typ vor dem Hintergrund absolut intakter Sensibilität), das Vorliegen angeborener Anomalien und Knochendeformitäten. Die angeborene Werdnig-Hoffmann-Amyotrophie kann von einem Neonatologen diagnostiziert werden. Differenzialdiagnose durchgeführt bei Myopathien, progressiver Duchenne-Muskeldystrophie, amyotropher Lateralsklerose, Syringomyelie, Poliomyelitis, Floppy-Child-Syndrom, Zerebralparese, Stoffwechselerkrankungen.
Um die Diagnose zu bestätigen, wird eine Elektroneuromyographie durchgeführt – eine Untersuchung des neuromuskulären Systems, dank derer charakteristische Veränderungen aufgedeckt werden, die eine primär muskuläre Läsion ausschließen und auf eine Pathologie des Motoneurons hinweisen. Biochemische Analyse Das Blut zeigt keinen signifikanten Anstieg der Kreatinphosphokinase, der für eine progressive Muskeldystrophie charakteristisch ist. MRT- oder CT-Scan der Wirbelsäule in seltenen Fällen visualisieren atrophische Veränderungen Vorderhörner des Rückenmarks, ermöglichen aber den Ausschluss anderer Wirbelsäulenpathologien (Hämatomyelie, Myelitis, Zyste und Tumor des Rückenmarks).
Die endgültige Diagnose einer Werdnig-Hoffmann-Amyotrophie wird nach Erhalt von Muskelbiopsiedaten und genetischen Studien gestellt. Morphologische Studie Die Muskelbiopsie zeigt eine pathognomonische faszikuläre Atrophie der Muskelfasern mit abwechselnden Zonen von Myofibrillenatrophie und unverändertem Muskelgewebe, das Vorhandensein einzelner hypertrophierter Myofibrillen und Bereiche mit Bindegewebswachstum. Die von Genetikern durchgeführte DNA-Analyse umfasst direkte und indirekte Diagnostik. Mit Hilfe direkte Methode Es ist auch möglich, den heterozygoten Träger einer Genaberration zu diagnostizieren, was für die genetische Beratung von Geschwistern (Brüdern und Schwestern) erkrankter Personen wichtig ist. Ehepaare Schwangerschaft planen. Dabei spielt eine quantitative Analyse der Anzahl der Gene am SMA-Locus eine wichtige Rolle.
Pränatale DNA-Tests können die Wahrscheinlichkeit verringern, ein Kind mit Werdnig-Hoffmann-Krankheit zu bekommen. Um jedoch fötales DNA-Material zu gewinnen, ist es notwendig, invasive Methoden der Pränataldiagnostik anzuwenden: Amniozentese, Chorionzottenbiopsie, Cordozentese. Eine im Uterus diagnostizierte Werdnig-Hoffmann-Amyotrophie ist eine Indikation für einen künstlichen Schwangerschaftsabbruch.
Behandlung der Werdnig-Hoffmann-Amyotrophie
Eine ätiopathogenetische Therapie wurde nicht entwickelt. Derzeit wird die Werdnig-Hoffmann-Amyotrophie durch eine Verbesserung des peripheren Stoffwechsels behandelt nervöses System und Muskelgewebe, um das Fortschreiten der Symptome zu verlangsamen. In der Therapie werden Kombinationen verschiedener Medikamente eingesetzt pharmakologische Gruppen: Neurometaboliten (Präparate auf der Basis von Schweinehirnhydrolysat, B-Vitaminen, Gamma-Aminobuttersäure, Piracetam), erleichtern die neuromuskuläre Übertragung (Galantamin, Sanguinarin, Neostigmin, Ipidacrin), verbessern den Myofibrillentrophismus (Glutaminsäure, Coenzym Q10, L-Carnitin, Methionin) , die die Durchblutung verbessern (Nikotinsäure, Scopolamin). Physiotherapie und Kindermassage werden empfohlen.
Moderne technologische Entwicklungen haben das Leben von Patienten und ihren Angehörigen durch den Einsatz automatisierter Rollstühle und tragbarer Beatmungsgeräte etwas erleichtert. Trägt zur Verbesserung der Patientenmobilität bei verschiedene Methoden orthopädische Korrektur. Die Hauptaussichten bei der Behandlung von SMA hängen jedoch mit der Entwicklung der Genetik und der Suche nach Möglichkeiten zusammen, genetische Aberrationen mithilfe von Methoden zu korrigieren Gentechnik.
Vorhersage
Die angeborene Werdnig-Hoffmann-Amyotrophie hat eine äußerst ungünstige Prognose. Wenn es sich in den ersten Lebenstagen eines Kindes manifestiert, tritt sein Tod in der Regel vor dem 6. Lebensmonat ein. Wenn die Klinik nach 3 Lebensmonaten beginnt, tritt der Tod im Durchschnitt im Alter von 2 Jahren ein, manchmal im Alter von 7–8 Jahren. Die frühkindliche Form zeichnet sich durch einen langsameren Verlauf aus, Kinder sterben im Alter von 14-15 Jahren.
Krankheitshäufigkeit
SMA ist eine der häufigsten seltenen Krankheiten und betrifft jedes 6.000–10.000 Neugeborene.
Ursache von SMA
SMA ist eine Erbkrankheit und geht mit Mutationen im SMN1-Gen einher.
Damit sich die Krankheit manifestiert, müssen beide Elternteile Träger einer Mutation in diesem Gen sein. Etwa jeder 40. Mensch trägt das rezessive SMA-Gen. Die Wahrscheinlichkeit, ein krankes Kind von zwei Trägern zu bekommen, liegt bei 25 %, mit der gleichen Wahrscheinlichkeit wird ein Kind von zwei Trägern keinen Gendefekt haben. In weiteren 50 % der Fälle ist er Träger von SMA, erkrankt aber selbst nicht.
In seltenen Fällen (weniger als 2 %) werden betroffene Kinder in Familien geboren, in denen nur ein Elternteil Überträger ist. Beim zweiten Elternteil kommt es bei der Ei- oder Spermienablage zu einer Genmutation.
Was wird durch die Mutation geschädigt?
Aufgrund eines defekten Gens im Körper ist die Produktion des SMN-Proteins, dem Überlebensprotein der Motoneuronen, gestört. Ohne dieses Protein sterben Motoneuronen – die Nervenzellen des Rückenmarks, die für die Bewegungskoordination und den Muskeltonus verantwortlich sind – ab, das Signal gelangt nicht zu den Muskeln der Beine, des Rückens und teilweise der Arme.
Ohne den nötigen Tonus verkümmern die Muskeln allmählich. Das Fehlen der Bauch- und Rückenmuskulatur führt unter anderem zu starken Verkrümmungen der Wirbelsäule und diese führen zu Atemproblemen, die bereits durch eine schwache Muskulatur vorliegen.
Die Krankheit kann sich bereits in den ersten Lebensmonaten oder in einem späteren Alter manifestieren.

Was bestimmt die Schwere der Erkrankung?
Für die Produktion des SMN-Proteins sind zwei Gene verantwortlich: SMN1 und SMN2.
Gleichzeitig ist SMN1 der Haupt-„Kunde“ dieses Proteins und SMN2 ist ein zusätzlicher Kunde, der Protein in einer Menge produziert, die für die normale Funktion des Körpers nicht ausreicht. Fehlt SNM1 im menschlichen Genom, übernimmt SNM2 Ersatzfunktionen, kann den Mangel jedoch nie vollständig ausgleichen.
Es gibt bis zu acht Kopien von SMN2 im Genom. Die Schwere der Erkrankung des Patienten hängt von der Anzahl der SMN2-Kopien ab, die eine Person besitzt. Solch komplexer Mechanismus Die Krankheit führt dazu, dass SMA mehrere Formen hat und der Zustand der Patienten sehr unterschiedlich ist.
Welche Formen von SMA gibt es?
Es gibt 4 Arten von SMA, die sich im Schweregrad und im Alter, in dem die Krankheit erstmals auftritt, unterscheiden.
SMA I, Werdnig-Hoffman-Krankheit. Die schwerste Form der Erkrankung manifestiert sich bei Säuglingen im Alter von 0 bis 6 Monaten. Kinder mit dieser Form haben von Geburt an Schwierigkeiten beim Atmen, Saugen und Schlucken und beherrschen auch die einfachsten kontrollierten Bewegungen nicht – halten Sie den Kopf nicht hoch, sitzen Sie nicht selbstständig. Bisher ging man davon aus, dass die Mehrheit (80 %) nicht älter als zwei Jahre wurde. Dank neuer Beatmungsstrategien und Sondenernährung Die Lebensdauer kann um einige weitere Monate verlängert werden.
SMA II, Dubowitz-Krankheit. Die ersten Manifestationen der Krankheit treten im Alter von 7 bis 18 Monaten auf. Eine Person mit dieser Art von SMA kann essen und sitzen, aber nicht selbstständig gehen. Die Lebenserwartung hängt vom Grad der Schädigung der Atemmuskulatur ab.
SMA III, Kugelberg-Welander-Krankheit. Die Krankheit tritt erstmals nach anderthalb Jahren auf. Solche Patienten können stehen (mit Schmerzen), aber nicht gehen. Für die Lebensdauer von SMA Typ III Dies hat in der Regel keinen Einfluss auf die Qualität, verschlechtert sie jedoch erheblich.
SMAIV, dieser Typ wird auch „adulte SMA“ genannt, da die Krankheit meist erst nach dem 35. Lebensjahr auftritt.
Symptome – Muskelschwäche, Skoliose und Zittern. Darüber hinaus kommt es zu Gelenkkontrakturen (eingeschränkte Beweglichkeit der Gelenke) und Stoffwechselstörungen.
Das Fortschreiten der Krankheit ist nicht sehr schnell, zunächst betrifft die Muskelschwäche die Beinmuskulatur, dann die Armmuskulatur. Normalerweise Probleme beim Schlucken und Atemfunktion Patienten nicht.
Die meisten Patienten mit SMA Typ IV können laufen, nur wenige müssen auf Rollstühle zurückgreifen.
SMA im Zusammenhang mit einer Störung des SMN-Gens, in medizinische Literatur genannt proximal – sie machen 95 % aller spinalen Amyotrophien aus. Es gibt eine ganze Reihe von SMAs, die nicht mit dem SMN-Gen assoziiert sind, aber sie sind selten. Hierzu zählt beispielsweise die Kennedy-Krankheit. Studien in den 1990er Jahren zeigten, dass die Kennedy-Krankheit nicht mit einem Abbau des SMN1-Gens einhergeht, sondern mit anderen genetischen Mutationen, die zu einer beeinträchtigten Aufnahme des SMN-Proteins führen. Die Krankheit manifestiert sich bei Menschen über 35 Jahren. SMBA ist vor allem durch eine Schwäche der Gliedmaßen gekennzeichnet.
Eine Art von SMA, die nicht mit dem SMN-Gen assoziiert ist, wird als SMA bezeichnet Kennedy-Krankheit. Die Tatsache, dass diese Krankheit manchmal immer noch als SMA bezeichnet wird, ist ein Anachronismus. Ende der 1960er Jahre, als es fertiggestellt wurde detaillierte Beschreibung Bei dieser Atrophie wurde sie als eine Form der SMA angesehen, da sie die gleichen Nerven und Muskeln betrifft wie bei drei Typen SMA (aber in viel geringerem Maße).
Wie wird es behandelt?
Derzeit gibt es keine radikale Behandlung für SMA.
Der internationale Konzern Biogen entwickelte das Medikament Spinraza, das den Zustand der Patienten, bei denen es während der Tests eingesetzt wurde, deutlich verbesserte. Derzeit ist das Medikament in den USA zugelassen; in Europa werden die geschätzten Kosten für einen jährlichen Kurs nach Schätzungen des Unternehmens etwa 270.000 Euro betragen, in Russland ist das Medikament nicht zertifiziert. Lebenslange Behandlung.
Kann man Patienten mit SMA helfen und wie genau?
Es ist noch nicht möglich, die Krankheit zu heilen, aber es ist möglich, den Zustand von Patienten mit SMA zu lindern verschiedene Wege kompensieren die Krankheitssymptome.
Bei schweren Formen der SMA muss den Patienten beim Atmen und Schlucken geholfen werden. Deshalb brauchen sie unbedingt Mobiltelefone. Ventilatoren, Hustensauger, Ambu-Beutel.
Kinder mit SMA brauchen auch unbedingt die Hilfe von Freiwilligen, die zumindest helfen können eine kurze Zeit Eltern ersetzen.
Kinder mit SMA können jederzeit Hilfe benötigen, daher sind Mütter und Väter immer auf der Hut und beherrschen selbst die notwendigen Reanimationsfähigkeiten für den Fall, dass das Kind plötzlich nicht mehr atmet.
Weniger schwer erkrankte Patienten benötigen Medikamente, die das Atmen erleichtern, Korsetts, Kinderwagen und andere Hilfsmittel, die Menschen mit schwacher Muskulatur die Bewegung und das Leben erleichtern.
Eine Krankheit, die über viele Jahre andauert, ist anstrengend, daher benötigen Patienten, insbesondere Erwachsene, häufig die Hilfe eines Psychologen.

Der Fonds ist in ganz Russland tätig. Die Arbeit der Stiftung hat zwei Hauptrichtungen – die Unterstützung von SMA-Patienten selbst und ihren Angehörigen und die Arbeit an systemischen Veränderungen in der Situation mit SMA in Russland.
Sie können die Aktivitäten der Stiftung durch eine Spende auf jede für Sie passende Weise unterstützen. Sie können helfen, indem Sie auf einer Sonderseite des Fonds einmalig oder regelmäßig spenden oder eine SMS an die Kurznummer 3443 mit dem Wort SMA und, durch ein Leerzeichen getrennt, dem Spendenbetrag senden – zum Beispiel SMA 300.
Kann man durch Impfungen an SMA erkranken?
In Europa und den USA konnte der Zusammenhang zwischen Impfungen und der Manifestation der Krankheit nicht nachgewiesen werden.
Das Verständnis, ob ein Zusammenhang zwischen SMA und Impfungen besteht, könnte helfen, den Unterschied zwischen SMA und Polio zu erklären. Poliomyelitis – Infektion wenn der Körper durch eine Infektion zunächst geschädigt wird gesundes Kind. Ein Kind mit SMA, das mit einem geschädigten Genom geboren wurde, mag äußerlich gesund aussehen, aber in Wirklichkeit ist es bereits krank, die Symptome seiner Krankheit treten einfach allmählich auf. In dieser Hinsicht handelt es sich bei SMA um dieselbe „verzögerte“ Erkrankung wie beispielsweise die Duchenne-Muskeldystrophie oder das Rett-Syndrom, bei der ein Kind, das sich seit einiger Zeit normal entwickelt, zuvor erworbene Fähigkeiten verliert und behindert wird.
Die meisten Manifestationen von SMA sind mit der Entwicklung erster motorischer Fähigkeiten verbunden. Die ersten Manifestationen der Krankheit fallen zeitlich mit mehreren altersbedingten Impfungen zusammen. Infolgedessen können eine Person und ihre Familie behaupten, dass sie „durch den Impfstoff krank geworden“ ist, in Wirklichkeit jedoch lediglich Anzeichen einer bereits bestehenden Krankheit gezeigt haben.
Wie wird festgestellt, dass ein Kind an SMA und nicht an einer anderen Krankheit leidet?
Obwohl SMA Anfang der 1890er Jahre erstmals vom österreichischen Neurologen Guido Werdnig und dem deutschen Neurologen Johann Hoffmann beschrieben wurde, wurde die Natur der Krankheit erst Ende des 20. Jahrhunderts vollständig verstanden. Das SMN1-Gen wurde 1995 entdeckt. Um die Diagnose SMA zu bestätigen, ist ein Gentest erforderlich.
In Russland wurden entsprechende Gentests Anfang der 2000er Jahre verfügbar. Ein Gentest auf SMA kann im Rahmen der gesetzlichen Krankenversicherung durchgeführt werden, in der Praxis kennen jedoch nicht viele Ärzte diese seltene Diagnose und überweisen Patienten zur entsprechenden Untersuchung. Die Kosten für eine solche Prüfung betragen kommerzielle Labore Moskau - etwa 6 Tausend Rubel.
Das Fehlen einer spezifischen Diagnostik hat auch zu einer Verwirrung bei den Diagnosen geführt. Die Mehrzahl der Patienten mit SMA in Russland ist nicht identifiziert; bei vielen der identifizierten Patienten ist die Diagnose „Werdnig-Hoffmann-Krankheit“ eingetragen, obwohl nicht alle von ihnen (insbesondere Erwachsene) tatsächlich an dieser Art von Krankheit leiden.
Wie viele Patienten mit SMA gibt es in Russland?

Das Medikament „Spinraza“, das den Zustand der Patienten deutlich verbessert. Foto von healthbeat.spectrumhealth.org
Unter Berücksichtigung der Häufigkeit der Erkrankung dürfte die Zahl der Patienten mit SMA in Russland zwischen sieben und vierundzwanzigtausend Menschen liegen. Heute sind etwa 400 Personen im Patientenregister der SMA Families Foundation eingetragen.
Wer in Russland hilft Menschen mit SMA und ihren Familien?
Gemeinnützige Stiftung „Vera“, Kinderhospiz „Haus mit Leuchtturm“, gemeinnützige Stiftung „Kinderpalliativ“, gemeinnützige Stiftung „Families of SMA“, Kinderpalliativdienst „Mercy“.
Seit 2014 entwickelt sich in Moskau ein gemeinsames Projekt des Dienstes „Mercy“ und der Stiftung „Families of SMA“ „SMA Clinics“. Bei Treffen, die einmal im Monat stattfinden, können sich Patienten von einem Lungenarzt, Orthopäden und Physiotherapeuten beraten lassen und Psychologe. IN In letzter Zeit Einige Treffen konzentrieren sich auch auf die Bedürfnisse erwachsener Patienten.
Berühmte Leute mit SMA

Die Italienerin Simona Spinoglio geboren mit Erbkrankheit– spinale Muskelatrophie Typ 2. Seit ihrer Geburt kann sie nicht mehr laufen und bewegt sich nur mit Hilfe eines Elektrorollstuhls. Aber ihr Leben ist erfüllt und ereignisreich; Nichts kann ihren Lebenswillen aufhalten.
Simone arbeitet für „ Hotline» Italienischer Verein „Families of SMA“ und hilft Kindern und Erwachsenen mit SMA und anderen neuromuskulären Erkrankungen.
Simone hat außerdem mehrere in der italienischen SMA-Community beliebte Lieder aufgenommen – über die Freiheit, trotz der Krankheit zu tun, was man will.
Russische Sängerin Julia Samoilova geboren in der Stadt Uchta (Republik Komi). Im Alter von zehn Jahren trat sie bei einem Benefizkonzert auf, danach wurde sie eingeladen, Gesang im örtlichen Pionierpalast zu studieren. Im Alter von fünfzehn Jahren begann sie ein Studium am städtischen Kulturhaus.
Im Jahr 2008 stellte sie ihr eigenes zusammen Musikgruppe(2010 aufgelöst). 2013 nahm sie am Wettbewerb „Faktor A“ des Fernsehsenders Rossija teil. Sie belegte den zweiten Platz und erhielt Alla Pugachevas persönliche Auszeichnung „Allas goldener Stern“. Im Jahr 2017 aufgrund des Ausschlusses Russlands Wettbewerbsprogramm konnte nicht am Eurovision Song Contest teilnehmen. Bewegt sich im Rollstuhl.
Programmierer von Vladimir Valery Spiridonov. Er schloss die Schule mit einer Goldmedaille ab und verteidigte anschließend seinen Ingenieurabschluss. Im Jahr 2015 plante Valery, an einem Experiment des italienischen Chirurgen Sergio Canavero zur Transplantation eines menschlichen Kopfes teilzunehmen (das Experiment wurde abgesagt).
Heute ist Valery Mitglied der öffentlichen Kammer der Stadt Wladimir und ein Experte für Themen zugängliche Umgebung, sowie der Schöpfer seiner eigenen Community „Desire for Life“, in der es um die Schaffung einer zugänglichen Umgebung und vielversprechende medizinische Projekte geht. Valery nimmt an vielen Fernsehprogrammen im russischen und ausländischen Fernsehen teil.
Was ist Werdnig-Hoffmann-Wirbelsäulenamyotrophie?
Spinale Amyotrophie von Werdnig-Hoffmann. Die Krankheit wurde 1891 von J. Werdnig und 1893 von J. Hoffmann beschrieben. Die Häufigkeit beträgt 1 pro 100.000 Einwohner, 7 pro 100.000 Neugeborene.
Was verursacht Werdnig-Hoffmann-Amyotrophie der Wirbelsäule?
Wird autosomal-rezessiv vererbt.
Pathogenese (was passiert?) während der Werdnig-Hoffmann-Amyotrophie der Wirbelsäule
Es wird eine Unterentwicklung der Zellen der Vorderhörner des Rückenmarks und eine Demyelinisierung der Vorderwurzeln festgestellt. Es gibt oft ähnliche Veränderungen in den motorischen Kernen und Wurzeln V, VI, VII, IX, X, XI und XII Hirnnerven. In der Skelettmuskulatur sind neurogene Veränderungen durch „Bündelatrophie“, einen Wechsel von atrophierten und intakten Muskelfaserbündeln sowie durch für primäre Myopathien typische Störungen (Hyalinose, Hypertrophie einzelner Muskelfasern, Hyperplasie) gekennzeichnet Bindegewebe).
Symptome einer spinalen Werdnig-Hoffmann-Amyotrophie
Es gibt drei Formen der Krankheit: angeborene, frühkindliche und späte, die sich im Zeitpunkt der Manifestation der ersten unterscheiden klinische Symptome und das Tempo des amyotrophen Prozesses.
Bei angeborene Form Ab den ersten Lebenstagen leiden Kinder unter einer generalisierten Muskelhypotonie und Muskelschwund sowie verminderten oder fehlenden Sehnenreflexen. Bulbarstörungen werden frühzeitig erkannt und äußern sich in trägem Saugen, schwachem Schreien, Zungenflimmern und vermindertem Rachenreflex. Die Krankheit geht mit osteoartikulären Deformitäten einher: Skoliose, Trichterbrust oder „Hühnerbrust“, Gelenkkontrakturen. Die Entwicklung statischer und motorischer Funktionen wird stark verlangsamt. Nur eine begrenzte Anzahl von Kindern entwickelt mit erheblicher Verzögerung die Fähigkeit, den Kopf hochzuhalten und selbstständig aufzusitzen. Allerdings bilden sich erworbene motorische Fähigkeiten schnell zurück. Viele Kinder mit einer angeborenen Form der Krankheit haben eine verminderte Intelligenz. Oft beobachtet Geburtsfehler Entwicklungen: angeborener Hydrozephalus, Kryptorchismus, Hämangiom, Hüftdysplasie, Klumpfuß usw.
Fließen. Die Krankheit verläuft schnell fortschreitend. Tod tritt vor dem 9. Lebensjahr auf. Eine der Haupttodesursachen sind schwere somatische Störungen (Herz-Kreislauf- und Atemversagen), die durch eine Schwäche der Brustmuskulatur und eine verminderte Beteiligung an der Atmungsphysiologie verursacht werden.
Bei frühkindliche Form Die ersten Krankheitszeichen treten in der Regel in der zweiten Lebenshälfte auf. Die motorische Entwicklung in den ersten Monaten ist zufriedenstellend. Kinder fangen an, rechtzeitig den Kopf zu heben, zu sitzen und manchmal auch aufzustehen. Die Krankheit entwickelt sich subakut, oft nach einer Infektion oder einer Nahrungsmittelvergiftung. Die schlaffe Parese ist zunächst in den Beinen lokalisiert und breitet sich dann schnell auf die Rumpf- und Armmuskulatur aus. Die diffuse Muskelatrophie geht mit Faszikulationen, Zungenflimmern, feinem Zittern der Finger und Sehnenkontrakturen einher. Muskeltonus, Sehnen- und Periostreflexe sind vermindert. In späteren Stadien kommt es zu einer generalisierten Muskelhypotonie und Symptomen einer Bulbarparese.
Fließen. Bösartig, wenn auch milder im Vergleich zur angeborenen Form. Der Tod tritt im Alter von 14 bis 15 Jahren ein.
Bei Spätform Die ersten Anzeichen der Krankheit treten im Alter von 1,5 bis 2,5 Jahren auf. In diesem Alter haben Kinder die Ausbildung statischer und motorischer Funktionen vollständig abgeschlossen. Die meisten Kinder gehen und rennen selbstständig. Die Krankheit beginnt unbemerkt. Bewegungen werden unbeholfen und unsicher. Kinder stolpern und fallen oft. Der Gang ändert sich: Sie gehen mit an den Knien angewinkelten Beinen (Gangart einer „Aufziehpuppe“). Die schlaffe Parese ist zunächst in den proximalen Muskelgruppen der unteren Extremitäten lokalisiert und breitet sich dann relativ langsam auf die proximalen Muskelgruppen der oberen Extremitäten und die Rumpfmuskulatur aus; Aufgrund der gut entwickelten subkutanen Fettschicht ist die Muskelatrophie normalerweise subtil. Typisch sind Faszikulationen, feines Zittern der Finger, Bulbussymptome – Flimmern und Atrophie der Zunge, verminderte Rachen- und Gaumenreflexe. Sehnen- und Periostreflexe klingen bereits ab frühe Stufen Krankheiten. Osteoartikuläre Deformitäten entwickeln sich parallel zur Grunderkrankung. Die ausgeprägteste Verformung der Brust.
Fließen. Bösartig, aber milder als die ersten beiden Formen. Eine Beeinträchtigung der Fähigkeit zum selbstständigen Gehen tritt im Alter von 10–12 Jahren auf. Die Patienten leben bis zu 20-30 Jahre.
Diagnose der spinalen Werdnig-Hoffmann-Amyotrophie
Die Diagnose basiert auf einer genealogischen Analyse (autosomal-rezessive Vererbung), klinischen Merkmalen (frühes Auftreten, Vorliegen einer diffusen Atrophie mit überwiegender Lokalisierung in den proximalen Muskelgruppen, generalisierter Muskelhypotonie, Faszikulationen und Flimmern der Zunge, Fehlen von Pseudohypertrophien, fortschreitend). und in den meisten Fällen bösartiger Verlauf usw.), die Ergebnisse der globalen (kutanen) und Nadelelektromyographie und der morphologischen Untersuchung der Skelettmuskulatur, die es uns ermöglichen, den Denervierungscharakter der Veränderungen zu erkennen.
Angeborene und frühe Formen sind vor allem von Erkrankungen zu unterscheiden, die zu den Syndromgruppen mit angeborener Muskelhypotonie („schlaffes Kind“-Syndrom) gehören: Oppenheim-Amyatonie, angeborene gutartige Form der Muskeldystrophie, atonische Form der Zerebralparese, erbliche Stoffwechselerkrankungen, chromosomale Syndrome usw. Die Spätform sollte von der spinalen Kugelberg-Welander-Amyotrophie, den progressiven Muskeldystrophien Duchenne, Erb-Roth usw. unterschieden werden.
Behandlung der spinalen Werdnig-Hoffmann-Amyotrophie
Bei Werdnig-Hoffmann-Wirbelsäulenamyotrophie werden Bewegungstherapie, Massage und Medikamente verschrieben, die den Trophismus des Nervengewebes verbessern – Cerebrolysin, Aminalon (Gammalon), Pyriditol (Encephabol).
An welche Ärzte sollten Sie sich wenden, wenn Sie an einer Werdnig-Hoffmann-Amyotrophie der Wirbelsäule leiden?
Neurologe
Aktionen und Sonderangebote
Medizinische Nachrichten
14.11.2019
Experten sind sich einig, dass es notwendig sei, die öffentliche Aufmerksamkeit auf die Probleme zu lenken Herz-Kreislauf-Erkrankungen. Einige sind selten, fortschreitend und schwer zu diagnostizieren. Dazu gehört beispielsweise die Transthyretin-Amyloid-Kardiomyopathie
14.10.2019
Am 12., 13. und 14. Oktober veranstaltet Russland eine große gesellschaftliche Veranstaltung für kostenlose Blutgerinnungstests – den „INR-Tag“. Die Aktion ist gewidmet Welttag Kampf gegen Thrombose. 04.05.2019
Die Inzidenz von Keuchhusten in der Russischen Föderation stieg im Jahr 2018 (im Vergleich zu 2017) fast um das Zweifache, auch bei Kindern unter 14 Jahren. Gesamtzahl Die Zahl der registrierten Keuchhustenfälle im Zeitraum Januar bis Dezember stieg von 5.415 Fällen im Jahr 2017 auf 10.421 Fälle im gleichen Zeitraum im Jahr 2018. Die Häufigkeit von Keuchhusten ist seit 2008 stetig gestiegen...
Medizinische Artikel
Fast 5 % von allen bösartige Tumore stellen Sarkome dar. Sie sind äußerst aggressiv, breiten sich schnell hämatogen aus und neigen nach der Behandlung zu Rückfällen. Manche Sarkome entwickeln sich jahrelang, ohne irgendwelche Anzeichen zu zeigen ...
Viren schweben nicht nur in der Luft, sondern können auch auf Handläufen, Sitzen und anderen Oberflächen landen und dabei aktiv bleiben. Daher empfiehlt es sich, auf Reisen oder an öffentlichen Orten nicht nur die Kommunikation mit anderen Menschen auszuschließen, sondern auch zu vermeiden...
Zurückkehren gute Sicht Und sich für immer von Brille und Kontaktlinsen zu verabschieden, ist der Traum vieler Menschen. Jetzt kann es schnell und sicher Wirklichkeit werden. Neue Möglichkeiten Laserkorrektur Die Sicht wird durch die völlig berührungslose Femto-LASIK-Technik eröffnet.
Kosmetika zur Pflege unserer Haut und Haare sind möglicherweise nicht so sicher, wie wir denken
- Muskelschwäche: Unbeholfenheit beim Bewegen, Gangstörung (Tritt mit gebeugten Knien).
- Verminderter Muskeltonus.
- Volumenreduzierung der Gliedmaßen aufgrund der Unterentwicklung der Muskulatur.
- Zittern (kleines Zucken, Zittern) der Gliedmaßen.
- Verminderte Mimik.
- Schluckstörung – Ersticken beim Schlucken.
- Atemstörungen und dadurch häufige Bronchitis, Pneumonie (Entzündung). Lungengewebe). Es tritt aufgrund einer Störung der Atemmuskulatur auf, was zur Entwicklung einer Blutstauung in der Lunge führt. Dies ist ein Zustand, der die Entwicklung einer Entzündung begünstigt.
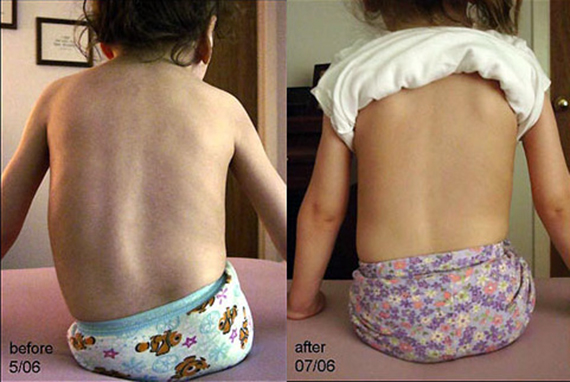
- Verformungen des Knochen- und Gelenkapparates: Brustkorb (Hühnerbrust, Trichterbrust), Wirbelsäule (Skoliose – Krümmung der Wirbelsäule).
- Irreversible Einschränkung der Beweglichkeit der Gliedmaßengelenke (Kontrakturen).
- Verlangsamung der körperlichen Entwicklung des Kindes.
Formen
- Angeboren - Anzeichen werden unmittelbar nach der Geburt beobachtet:
- das Kind ist lethargisch;
- nimmt die Brust nicht gut an;
- schwacher Schrei;
- reagiert träge auf die Umwelt;
- Diese Form geht häufig mit Anzeichen einer Skelettentwicklungsstörung einher (Skoliose – Krümmung der Wirbelsäule, Deformation der Brust).
- Früh Kinder: Das Kind entwickelt sich bis zum sechsten Monat normal, dann (häufig danach). Darminfektion) kommt es zunächst zu Lähmungen in den Beinen, dann in den Armen und der Muskeltonus nimmt ab. Kinder halten ihren Kopf nicht hoch, setzen sich nicht hin und drehen sich nicht um.
- Spät: Die ersten Anzeichen treten im Alter von 1–3 Jahren auf, oft bleibt das Auftreten der ersten Anzeichen unbemerkt. Das Kind entwickelt sich normal, dann beginnt der Gang zu stören, Bewegungen werden unbeholfen, dann kommt es zu einer allgemeinen Abnahme des Muskeltonus.
Ursachen
- Die Krankheit ist erblich, das heißt, sie wird von den Eltern auf die Kinder übertragen.
Diagnose
Wenn die oben beschriebenen Symptome auftreten, sollten Sie sich an uns wenden.
Der Arzt führt eine vollständige Untersuchung des Kindes durch, führt eine neurologische Untersuchung durch und bespricht mit den Eltern auch die Reihenfolge und den Zeitpunkt der Entwicklung von Krankheitszeichen.
Es ist wichtig, Daten über das mögliche Vorliegen von Anzeichen der Krankheit bei einem Familienmitglied zu sammeln, die den Arzt möglicherweise dazu veranlassen, über die erbliche Natur der Krankheit nachzudenken.
Zur Bestätigung der Diagnose verschreibt der Arzt:
- ENMG (Elektroneuromyographie): Sensoren auf der Haut zeichnen den Durchgang eines Nervenimpulses auf. Mit der Methode können Sie feststellen, was Bewegungsstörungen verursacht;
- Muskelbiopsie: Ein kleiner Teil des Muskels wird zur Untersuchung mit einer Nadel entnommen;
- MRT (Magnetresonanztomographie) des Kopfes: um andere Pathologien auszuschließen (mit der Methode können Sie die Struktur jedes Körperteils Schicht für Schicht untersuchen);
- Die genetische Analyse (Untersuchung der Chromosomenstruktur, Suche nach genetischen Mutationen) ist eine Methode, die die Diagnose bestätigt.
Behandlung der Amyotrophie der Wirbelsäule bei Kindern Werdnig-Hoffmann
Derzeit gibt es keine Methoden oder Medikamente, die die Krankheitsursache beeinflussen.
Zur Verbesserung der Ernährung des Nervensystems werden Methoden eingesetzt:
- Vitamintherapie;
- Nootropika (Medikamente, die die Ernährung des Gehirns verbessern);
- Massage;
- Physiotherapie.
Komplikationen und Folgen
- Unbeweglichkeit.
- Atemstillstand.
- Lebensgefahr.
Fließen frühe Form milder: Der Tod tritt im Alter von 14-15 Jahren ein.
In der Spätform verlieren Kinder im Alter von 10–12 Jahren die Gehfähigkeit und werden 20–30 Jahre alt.
Prävention der spinalen Amyotrophie im Kindesalter Werdnig-Hoffmann
- Aufgrund der erblichen Natur der Krankheit ist eine Vorbeugung nicht möglich.